10 Tumoren der Niere
Werbung

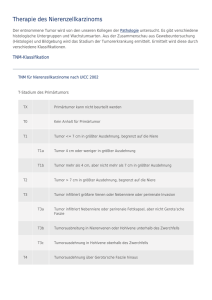
10 Tumoren der Niere 9.3 Multiple endokrine Neoplasien (MEN) DEFINITION Autosomal-dominant vererbte Erkrankungen Tab. 9.4 MEN-Syndrome Syndrom Konstellation MEN I (WermerSyndrom) •Nebenschilddrüsenadenom (95 %) mit primärem Hyperparathyreoidismus (pHPT) •endokrine Pankreastumoren (50 %): am häufigsten Insulinom, Gastrinom •Hypophysentumoren (30 %): Prolaktinom, Akromegalie, Morbus Cushing MEN IIa (SippleSyndrom) •medulläres Schilddrüsenkarzinom (100 %) •Phäochromozytom (50 %) •Nebenschilddrüsenadenom (20 %) mit pHPT MEN IIb (GorlinSyndrom) zusätzlich zu MEN IIa: •Schleimhautneurinome an der Zunge und gastrointestinal (schmerzhafte „Knötchen“ an der Zunge, Megakolon) •marfanoider Habitus familiäres medulläres Schilddrüsenkarzinom (FMTConly) •nur medulläres Schilddrüsenkarzinom mit charakterischen Kombinationen verschiedener neuroendokriner Tumoren. Epidemiologie: Die Häufigkeit von MEN I und MEN II beträgt jeweils 1: 50000. Der Typ MEN IIa (70 %) ist wesentlich häufiger als der Typ MEN IIb (10 %) und das familiäre medulläre Schilddrüsenkarzinom (20 %). Ätiologie: Die Syndrome werden autosomal-dominant vererbt (Familienanamnese!). Sporadische Fälle erklären sich durch Neumutationen. Bei MEN I ist das Menin-Gen (Tumorsuppressorgen), bei MEN II das Ret-Protoonkogen mutiert. Einteilung: Siehe Tab. 9.4. Therapie und Prophylaxe: Die Tumoren werden operativ entfernt. Prophylaktische Maßnahmen sind eine genetische Diagnostik und Beratung von Familienangehörigen, regelmäßige Vorsogeuntersuchungen bei Mutationsträgern und eine Thyreoidektomie bei MEN II. 10 Tumoren der Niere 10.1 Benigne Nierentumoren Gutartige Nierentumoren sind selten (Tab. 10.1). 10.2 Maligne Nierentumoren 10.2.1 Nierenzellkarzinom Synonym: Hypernephrom, Grawitz-Tumor DEFINITION Parenchymatöser Nierentumor, der von rena- len Tubulusepithelien ausgeht. Epidemiologie: Nierenzellkarzinome machen mit einer Inzidenz von 8/100000 Einwohner/Jahr 3 % aller Malignome bei Erwachsenen aus (m:w = 2:1) und treten gehäuft nach dem 50. Lebensjahr auf. Familiäre Formen entwickeln sich häufig bereits im 20.-40. Lebensjahr. Ätiologie: Zu den Risikofaktoren zählen Rauchen, Analgetikaabusus (Analgetikanephropathie; s. Niere S. 383), eiweißreiche Ernährung, Adipositas, berufliche Schadstoffe (Asbest, Kadmium, Blei, Trichlorethylen) und erworbene Nierenzysten bei Dialysepatienten. Das früher verwendete Röntgenkontrastmittel Thorotrast spielt heute keine Rolle mehr. 5 % d. F. treten familiär gehäuft auf (chromosomale Aberrationen, von-Hippel-Lindau-Syndrom). PATHO Klinische Pathologie: Das Nierenzellkarzinom geht von den renalen Tubulusepithelien aus und ist bei Diagnose stellung meist schon 3–15 cm groß. Er entsteht meistens Das klarzellige Nierenzellkarzinom erinnert histologisch stark an Ne- bennierenrindenzellen (→ früher gebräuchliche Bezeichnung Hyper- nephrom). im Bereich des Nierenpols. Im Frühstadium imponiert es als gut differenzierte Gewebswucherung und ist von einer dicken Pseudokapsel umgeben (→ einfache Tumorresektion und gute Prognose). Im Verlauf wölbt sich der Tumor am Nierenpol vor oder destruiert große Anteile des Nierenparenchyms. Dabei bricht es in die Capsula adiposa, das Nierenbecken oder das Gefäßsystem ein. Makroskopisch zeigen sich Blutungen, Verkalkungen, Narben, Regressionszysten und Nekrosen. Histologisch handelt es sich um Adenokarzinome, bei denen verschiedene Typen (Tab. 10.2) unterschieden werden. Klinik: Im Frühstadium sind die Patienten meist asymptomatisch. Schmerzlose Makrohämaturie und kolikartige Flankenschmerzen sind die häufigsten Erstsymptome, entwickeln sich aber erst mit Einbruch des Tumors in das Sammelrohrsystem. Nicht selten bricht der Tumor in die linke V. renalis (→ im Liegen persistierende Varikozele des linken Hodens durch Behinderung des venösen Abstroms aus der linken V. testicularis und dem Plexus pampiniformis) oder die V. cava inferior ein (→ Kavathrombose mit Emboliegefahr). Palpabler Tumor, Gewichtsverlust und intermittierendes Fieber kennzeichnen fortgeschrittene Tumorstadien. Häufige paraneoplastische Syndrome sind Hyperkalzämie (durch PTHrP-Produktion), Hypertonie (durch Reninproduktion), Polyglo- Heruntergeladen von: Thieme E-Books & E-Journals. Urheberrechtlich geschützt. 644 645 Herz/Kreislauf 10.2 Maligne Nierentumoren klinische Pathologie Histologie Besonderheiten Angiomyolipom (häufigster benigner mesenchymaler Nierentumor) Mischtumor aus Fett, glatten Muskelzellen und dickwandigen Gefäßen, ausgehend von perivaskulären epitheloiden Zellen Makroskopie: häufig multiple Einblutungen, Nekrosen, Kalkeinlagerungen „buntes Bild“, proliferierendes Fett- und Muskelgewebe, Blutgefäße, Kernatypien in 50 % d. F. familiär, z. B. bei tuberöser Sklerose bei Verdrängungssymptomen (selten) operative Entfernung sehr selten maligne Entartung Nierenzelladenom (Zufallsbefund in 10 % aller Autopsien) ausgehend vom Nierenparenchym (Nephron- und Sammelgangsystem), keine Malignitätszeichen, einzeln oder multipel, häufig in der Nierenrinde Makroskopie: gelbe, bohnengroße Knötchen mit scharf begrenztem Rand monomorphe Zellen ohne Zellatypien, häufig Expression von Vimentin, Keratin und Antigenen des jeweiligen Tubussegments, Differenzierung in papillären, onkozytären oder metanephrogenen Typ •papillärer und onkozytärer Typ: meistens asymptomatisch •metanephrogener Typ: häufig Hypertonie, Polyzythämie und Hämaturie → bei Symptomen Entfernung Nierenonkozytom ausgehend vom Nierenepithel Makroskopie: rotbraune Schnittfläche mit zentraler, sternförmiger Narbe große Zellen mit granulärem, eosinophilem Zytoplasma langsames Wachstum, Durchmesser > 10 cm möglich Nierenkapseltumor („Kapsulom“, Zufallsbefund in 10 % aller Autopsien) ausgehend von pluripotenten Nierenblasten der Nierenkapsel Leiomyome, Fibrome, Lipome oder Mischtumoren keine Therapie notwendig renomedullärer Interstitialzelltumor (oft Zufallsbefund) Tumor der Markpyramiden, ausgehend von Interstitiumzellen Makroskopie: kleine, gräuliche Knötchen sehr kollagenfaserreich, Amyloidablagerungen im Tumorzentrum Prostaglandinproduktion ohne Beziehung zur Blutdruckregulation meist keine Therapie notwendig Juxtaglomerularzelltumor (sehr selten) ausgehend von juxtaglomerulären Zellen Makroskopie: grau-weiße bis gelbliche Schnittfläche uniforme, runde bis spindelzellige, eosinophile Zellen mit intrazytoplasmatischen Reninkörnchen Erkrankungsgipfel um das 20. Lebensjahr, mehr Frauen betroffen Reninproduktion → Hypertonie, Hypokaliämie und Hyperaldosteronismus Therapie: Tumorexstirpation klarzelliges Karzinom 75 % (häufigster Typ) •ausgehend von den proximalen Tubuluszellen •große Zellen mit transparentem, hellem Zytoplasma (hoher Glykogen- und Lipidgehalt) und scharf begrenzten Zellgrenzen („pflanzenzellartig“) •meist solides Wachstumsmuster papilläres Karzinom (Abb. 10.1) 10 % •ausgehend von den proximalen Tubuluszellen •kubische oder eosinophil-zylindrische Zellen, die verzweigten Bindegewebsstilen aufsitzen chromophober Typ 5% •ausgehend von den Schaltzellen des Sammelrohrs •voluminöse Zellen mit nichttransparentem, feinretikulärem Zytoplasma und zahlreichen Mikrovesikeln (Anfärbung mit kolloidalem Eisen) Sammelgangtyp 1 % (schlechteste Prognose) •ausgehend von den Sammelrohrzellen •tubuläre Strukturen, Desmoplasie des Tumorstromas, häufig sarkomartige Areale mit Spindelzellen tubulomuzinöser Typ selten (sehr gute Prognose) •ausgehend vermutlich von Abschnitten des distalen Nephrons und dem Sammelrohr •charakteristische interstitielle Schleimablagerungen zwischen homogenen, trabekulären Epithelien transitionalzelliger Typ sehr selten •ausgehend von der zentralen Markregion (Bellini-Gänge) •Tumorzellen mit urothelialer Differenzierung neuroendokriner Typ sehr selten •Nachweis von Chromogranin A, Synaptophysin und NSE unklassifizierter Typ selten (schlechte Prognose) •Diagnose nur nach Ausschluss der übrigen Typen erlaubt •sarkomartiges und mitosereiches Tumorgewebe •frühe Infiltration von Nachbarstrukturen Stauffer-Syndrom (Leberfunktionsstörung mit erhöhter AP). Immun/Rheuma Charakteristika Infektiologie Häufigkeit Neoplasien Typ Niere Endokrinium/Stoffw. Verdauung Heruntergeladen von: Thieme E-Books & E-Journals. Urheberrechtlich Atmung geschützt. Entartungstendenz (Adenom-KarzinomSequenz) Tab. 10.2 Histologische Typen des Nierenzellkarzinoms bulie (durch Erythropoetinproduktion), Cushing-Syndrom (durch ACTH-Produktion), das Lambert-Eaton- (Antikörper gegen präsynaptische Kalziumkanäle) und das Blut Tumor Gefäße Tab. 10.1 Benigne Nierentumoren 646 10 Tumoren der Niere a [aus: Krams et al., MERKE Das klassische Warnsignal des Nierenzellkarzinoms ist die schmerzlose Hämaturie. Metastasierung: Hämatogen metastasiert das Nierenzellkarzinom in Lunge, Mediastinum, Leber, Gehirn und Skelett. Die lymphogene Ausbreitung (seltener) erfolgt primär über die Lymphknoten des Nierenhilus sowie die paraaortalen und parakavalen Lymphknoten. RADIO Diagnostik: Eine Hämaturie (insbesondere ohne Zeichen einer Harnwegsinfektion) ist stets verdächtig und sollte eine umfassendere Diagnostik nach sich ziehen. Sonografisch (Abb. 10.2a) imponiert das Nierenzellkarzinom als solider Tumor mit ausgeprägter Konturvorwölbung und inhomogener Binnenstruktur ohne dorsale Schallverstärkung. Häufig sind zystische Einschmelzungen, Verkalkungen und eine Infiltration von Nierenvenen und/oder V. cava nachweisbar. Die farbkodierte Duplexsonografie und die Angio-CT (Abb. 10.2 b und c) zeigen typische Hypervaskularisationen und können Tumorzapfen in der Nierenvene und/oder V. cava inferior darstellen. Mithilfe der i. v.-Pyelografie können das Ausmaß der tumorbedingten Gewebeverdrängung und ggf. ein Einbruch ins Nierenbecken beurteilt werden. Nach gesicherter Diagnose sollte zum weiteren Staging ein Röntgen Thorax, eine Oberbauchsonografie und bei klinischem Verdacht eine Skelettszinigrafie oder eine CCT durchgeführt werden. Häufige Laborbefunde sind eine Anämie (in 20–30 % d. F.) oder auch eine Polyglobulie, eine Leuko- und Thrombozytose, erhöhte Entzündungsparameter und pathologische Nieren- und Leberparameter. Im Urinstatus finden sich häufig eine Erythrozyturie und eine Proteinurie. Verlässliche Tumormarker sind nicht verfügbar. † b † å c Abb. 10.2 Nierenzellkarzinom. a Sonografisch echoarmer Tumor in der linken Niere. b und c Nierenzellkarzinom am rechten unteren Nierenpol mit Hypervaskularisation in der arteriellen Phase (b) und Hypodensität in der portalen Phase (c). [aus: Reiser, Kuhn, Debus, Duale Reihe Radiologie, Thieme, 2011] Tab. 10.3 TNM-Klassifikation des Nierenzellkarzinoms Stadium Ausdehnung T1 Tumor ≤ 7 cm, auf Niere begrenzt •T 1a: Tumor ≤ 4 cm •T 1b: Tumor 4–7 cm T2 Tumor ≥ 7 cm, auf Niere begrenzt T3 Tumor infiltriert Venen, Nebenniere oder perirenales Gewebe, Gerota-Faszie aber nicht durchbrochen: •T 3a: Tumor infiltriert Nebenniere oder perirenales Gewebe •T 3b: Tumor infiltriert makroskopisch Nierenvenen oder V. cava unterhalb des Zwerchfells •T 3c: Tumor infiltriert V. cava auch oberhalb des Zwerchfells T4 Tumor durchbricht Gerota-Faszie N0 keine Lymphknotenmetastasen N1 solitäre regionale Lymphknotenmetastasen Stadieneinteilung: Siehe Tab. 10.3 und Tab. 10.4. Differenzialdiagnosen: Infrage kommen alle Ursachen einer Hämaturie (s. Leitsymptome S. C 85). Insbesondere die klinische Abgrenzung zur Nephrolithiasis kann schwerfallen, da auch diese kolikartige Flankenschmerzen auslösen kann. Sonografisch sollten andere Raumforderungen wie Nierenzysten (scharf begrenzte, echofreie å N2 mehrere regionale Lymphknotenmetastasen M0 keine Fernmetastasen M1 Fernmetastasen Heruntergeladen von: Thieme E-Books & E-Journals. Urheberrechtlich geschützt. Abb. 10.1 Papilläres Nierenzellkarzinom. Kurzlehrbuch Pathologie, Thieme, 2010] Rundherde mit dorsaler Schallverstärkung), Hämatome (heterogene, echoreiche und -arme, unscharf begrenzte Raumforderung), benigne Tumoren und Metastasen ausgeschlossen werden. MERKE Das Nierenzellkarzinom spricht sehr schlecht auf Prognose: Die 5-Jahres-Überlebensrate nach Radikaloperation hängt stark vom Stadium zum Diagnosezeitpunkt ab (s. Tab. 10.4). I Tumor auf Niere begrenzt (T 1 N0 M0) 70–85 % von Fernmetastasen die Therapie der Wahl bei Nierenzellkarzinom. II Infiltration von perirenalem Fettgewebe (T 2 N0 M0) 50–65 % III Infiltration von V. renalis und/oder V. cava und/oder Lymphknoten (T 1–3 N0–1 M0) N0: 25–50 % N1: 5–15 % IV Infiltration benachbarter Organe oder Fernmetastasen (T 4 N0 M0 oder jedes T M1) ≤5% 10.2.2 Nephroblastom Siehe Pädiatrie S. B 581. 11 Tumoren in bestimmten Kompartimenten 11.1 Tumoren des Mediastinums Epidemiologie: Mediastinaltumoren sind insgesamt selten. Am häufigsten sind Lymphome, Thymome (ca. 20 % d. F., mittleres Erkennungsalter 40. Lebensjahr) und neurogene Tumoren. Einteilung: Die Tumoren können von verschiedenen mediastinalen Strukturen ausgehen und werden nach ihrer Lokalisation eingeteilt (Tab. 11.1). Thymome (ca. 20 % aller Mediastinaltumoren, mittleres Erkrankungsalter: 40. Lebensjahr): 75 % der Tumoren sind primär benigne, 25 % maligne (= Thymuskarzinom). Da gutartige Thymome aber häufig entarten, werden sie generell als bösartig eingestuft. Die WHO unterscheidet abhängig vom Wachstumsverhalten folgende Typen: ▪ A-Thymome: abkapselte Tumoren ohne infiltratives Wachstum oder Metastasen ▪ B-Thymome: lokal infiltratives Wachstum, intrathorakale Metastasen ▪ C-Thymome: hochmaligne Karzinome mit infiltrativem Wachstum und Fernmetastasen. Makroskopisch imponieren Thymome als feste, gelbbraune, septierte Strukturen. In großen Tumoren finden sich häufig Zysten, Einblutungen und Nekrosen. MikroTab. 11.1 Tumoren des Mediastinums Lokalisation Mediastinaltumoren und -zysten vorderes Mediastinum •Thymome •Schildrüsentumoren •maligne Lymphome: Hodgkin- (v. a. nodulär-sklerosierender Typ) und Non-Hodgkin-Lymphome •Lymphknotenmetastasen (z. B. bei Bronchialkarzinom) •benigne Lymphome: angiofollikuläre Lymphknotenhyperplasie bzw. Morbus Castleman •Weichteilsarkome •Lipome •Teratome •Dermoide mittleres Mediastinum •Perikard-, Pleura- und bronchogene Zysten •Teratome •maligne Lymphome •Lymphknotenmetastasen •benigne Lymphome hinteres Mediastinum •neurogene Tumoren: Schwannome, Neurofibrome, Ganglioneurome, Neuroblastome •Ösophaguszysten und -tumoren Niere Endokrinium/Stoffw. Verdauung Heruntergeladen von: Thieme E-Books & E-Journals. Urheberrechtlich Atmung geschützt. MERKE Die radikale Nephrektomie ist bis zum Auftreten Immun/Rheuma 5-JahresÜberlebensrate Infektiologie Ausdehnung Blut Tab. 10.4 AJCC-Klassifikation und Prognose des Nierenzellkarzinoms Stadium Bei Metastasen kann eine Nephrektomie auch aus palliativen Gründen (Blutungen, Schmerzen) indiziert sein, eine Heilung ist allerdings äußerst selten möglich (< 0,8 % d. F.). Einzelne Lungen- oder Lebermetastasen können reseziert werden. Bei multiplen Metastasen kommen folgende Therapieansätze infrage: ▪ Immuntherapie mit Interferon-α und Interleukin-2 ▪ monoklonale VEGF-Antikörper Bevacizumab ▪ Tyrosinkinasehemmer wie Sunitinib ▪ mTOR-Inhibitor Temsirolimus. Gefäße eine Chemo- und Strahlentherapie an. Neoplasien Therapie: In den Stadien I–III ist eine radikale Nephrektomie indiziert (En-bloc-Entfernung von Niere, perirenaler Fettkapsel, Nebenniere, Harnleiter sowie der Vasa spermatica bzw. ovarica) mit Ausräumung der parakavalen und paraaortalen Lymphknoten und ggf. Entfernung eines Tumorzapfens aus der V. cava inferior. Bei kleinen Karzinomen (< 4 cm) kann eine Teilresektion der Niere erwogen werden 647 Herz/Kreislauf 11.1 Tumoren des Mediastinums