Gold Katalyse
Werbung

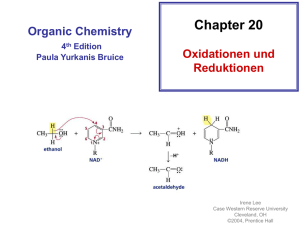
Gold Katalyse Literatur: Angew. Chem. Int. Ed. 2006, 45, 7896-7936 Homogene und Heterogene Hydrogenierung / Dehydrogenierung Heterogene Hydrogenierung • 1950, Couper und Eley : Gold-Oberflächen katalysieren die Umwandlung von para-Wasserstoff in ortho-Wasserstoff Gold-Oberflächen können H2 aktivieren • 1963, Erkelens, Kemball und Gawly: Erste durch Gold katalysierte Hydrogenierung von Alkenen Aktivierungsenergie für Hydrogenierung ist höher als für Dehydrogenierung Heterogene Hydrogenierung • 1973, Bond et al.: Effiziente Hydrogenierung bei niedrigen Temperaturen und geringem Goldanteil • Abhängigkeit vom Trägermaterial Heterogene Hydrogenierung • Chemoselektive Monohydrogenierung • D2 reagiert viel langsamer als H2 → Brechen der H-H Bindung ist geschwindigkeitsbestimmender Schritt Heterogene Dehydrogenierung / Isomerisierung • 1928, Clark und Toplet: • 1969, Inami et al.: • 1980, Satchler et al.: Eine Monoschicht Au auf Pt verbessert die Rate der Dehydrogenierung von Cyclohexen bei 100 °C um den Faktor 5 Homogene Hydrogenierung • 2006, Corma: Möglicher Mechanismus der homogenen Hydrogenierung Substitution Insertion Heterolytische Spaltung Addition Heterolyse + Eliminierung Oxidationsstufe von Au bleibt unverändert Heterolytische Spaltung geschwindigkeitsbestimmender Schritt Homogene Hydrogenierung • 1995, Baker et al.: Keine β–Wasserstoff Eliminierung bei Au-Komplexen Homogene Dehydrogenierung • 1999, Hosomi: Hexaalkyldistannane • 2005, Ito et al.: Dehydrogenierende Silylierung 1.Übersicht ∙ Trägerfixierte Gold-Katalysatoren → Epoxidierung von Alkenen → Oxidation von Alkoholen ∙ Heterogene Gold-Katalysatoren → Effiziente C-H-Aktivierung ∙ Legierungen von heterogenen Goldkatalysatoren mit Pd → direkte Bildung von Wasserstoffperoxid aus den Elementen 2.Epoxidierung ∙ Epoxidierung von Ethen mit Sauerstoff und Ag-Katalysator: Selektivität 90 % ∙ Oxidation von Propen problematisch: Selektivität für Epoxid < 10 % ∙ Lösung: Trägerfixierte Goldkatalysatoren für die Epoxidierung von Propen, H2 als Red ∙ verwendete heterogene Gold-Katalysatoren: - Au/TiOx - Au/TS-1 (Si : Ti von 500 : 1) → aktive Zentren: Gold-Spezies mit Durchmesser < 2nm - Au auf mesoporösen Titansilicat-Trägern → Selektivität > 90% zugunsten von Propenoxid, bei Umsatz von 7% ∙ katalytische Mengen Peroxid initiieren Oxidation von Alkenen mit O2 → sehr hohe Selektivitäten zugunsten des Epoxids für Cyclohexen, Styrol, cis-Stilben und Cycloocten ∙ Selektivität stark abhängig vom Lösungsmittel: am besten für substituierte Benzole ∙ „Grüne“ Chemie: - bestimmte Katalysatoren auch in Abwesenheit von Lösungsmitteln aktiv - niedrige Reaktionstemperaturen Tabelle 1: Oxidation von cis-Cycloocten mit O2 unter lösungsmittelfreien Bedingungen 3. Oxidation von Alkoholen und Aldehyden Abbildung 1: Oxidation unterschiedlicher Alkohole mit verschiedenen Katalysatoren ∙ bisher: trägerfixierte Pd-Nanopartikel - oxidieren Glucose → Gluconsäure Glycerin → Glycerinsäure aber: wenig selektiv ∙ Lösung: trägerfixierte Au-Nanopartikel - hohe Effizienz und Selektivität für Oxidation von Alkoholen und Diolen - Voraussetzung: Vorhandensein einer Base - Katalysatoren: Au/C - auch Oxidation von Zuckern möglich ∙ Au/Graphit katalyisiert Oxidation von Glycerin zu Glycerinsäure: - O2 als Ox, milde Bedingungen, NaOH → Selektivität bis zu 100%, Ausbeute 60 % - Selektivität abhängig vom Verhältnis NaOH/Glycerin - mit Pd/C bzw. Pt/C viele Nebenprodukte ∙ „nackte“ Au-Kolloidpartikel katalysieren: - Oxidation von Glucose zu Gluconsäure - Oxidation von 1,2-Diolen Schema 1: Mechanistisches Modell für die Glucose-Oxidation ∙ Au/CeO2: - Oxidationen: Alkohol → Aldehyd, bzw. Keton; Aldehyd → Carbonsäure - O2 als Ox, lösungsmittelfrei, milde Bedingungen → hohe Selektivität ∙ Legierung von Pd mit Au aus Au/TiO2-Kat: - effiziente Oxidation von primären Alkoholen und Diolen ∙ Goldkomplexe: - selektive Oxidation von Alkoholen zu Aldehyden und Ketonen - „grüne“ Chemie: Reaktion mit Luft, Umsatz bis 100%, Selektivität 99 % für Benzaldehyd 4. C-H-Aktivierung ∙ besonderes Interesse: aerobe Oxidation von Cyclohexan OH Au-Kat. O2 O + ∙ industrieller Prozess: 70-85% Ausbeute, Umsatz 4% ∙ Au-Katalysatoren: - Au/ZSM-5 → 90 % Selektivität für Cyclohexanon - Au/MCM-41 → > 90 % Selektivität für Cyclohexanon - Wiederverwendung der Katalysatoren problematisch → hohe Selektivitäten, allerdings bei hohen Temperaturen (140-160°C) ∙ Au/C-Katalysatoren: - Oxidation von Cyclohexan bei < 100°C - sehr hohe Selektivität bei niedrigem Umsatz - Tendenz identisch zu Pt/C und Pd/C: Selektivität nur vom Umsatz an Cyclohexan abhängig 5. Direkte Wasserstoffperoxidsynthese ∙ Industrie: Anthrachinonverfahren - problematisch für kleine Mengen ∙ optimal: Synthese aus den Elementen (wird seit 90 Jahren angestrebt) ∙ Au/Al2O3: - katalysiert direkte Reaktion von H2 und O2 - bei Au-Pd/Al2O3 Reaktion noch schneller, aber: Selektivitätsproblem → auch Hydrierung zu H2O und Zersetzung in H20 und O2 werden katalysiert Abbildung 2: Auswirkung von Additiven auf die H2O2 – Menge bei der direkten Oxidation von H2 ∙ Au/SiO2 bzw. Au-Pd/SiO2: - selbst bei 10°C noch aktiv ∙ Fe2O3 bzw. TiO2 als Trägermaterial: - Selektivitäten von bis zu > 95% (bei kurzen Reaktionszeiten) Tabelle 2: Einfluss der Reaktionszeit auf den H2-Umsatz und die Selektivität der H2O2-Synthese Nucleophile Additionen an p-Systeme Gold-Komplexe aktivieren für einen nucleophilen Angriff: • C-C-Doppel- und C-C-Dreifachbindungen in I. Isolierten Systemen II. Kumulierten Systemen III. Konjugierten Systemen • C-O Doppelbindungen in Carbonylgruppen Einfache Nucleophile ohne konjugierte Mehrfachbindungen WW mit dem p-System [Au] + [Au] R R R Nucleophile Addition R H + H+ Nu R - [Au] R Nu R + Nu[Au] R Protodemetallierung Diastereoselektive anti-Addition des Gold-Substituenten und des Nukleophils A) Alkine 1976: Alkine mit Tetrachloridogoldsäure (Thomas et al.) O 7 Mol-% H[AuCl4] MeOH/H2O 1985: Hydrochlorierung von Ethin (Hutchings) H trägerfixiertes AuIII HCl Cl A) Alkine Effiziente Synthese von Furanen durch (Z)-3-Ethinylallylalkoholen R1 R5 R4 HO R 3 0.1 Mol-% AuCl3 R5 R 3 R5 = H O R1 R2 R4 R4 O R2 R1 Spezifische Reaktion mit dem Alkin in Gegenwart des aktivierten Alkens Ph Ph O HO MeCN, RT COOMe O 5 Mol-% AuCl O COOMe 90 % R3 R2 B) Allene • Anspruchsvollste Substrate für Additionsreaktionen • Chemoselektivität, Diastereoselektivität und Regioselektivität sind relevant Cycloisomerisierung von Allenylcarbinolen zu 2,5-Dihydrofuranen HO O 10 Mol-% AuCl3 OTBS OTBS CH2Cl2, RT Addition von Anilinen an chirale Allene NHPh 10 Mol-% AuBr3 Ph 94 % ee PhNH2 Ph 88 % ee C) Alkene Zwei denkbare Reaktionswege: I. Aktivierung des Alkens und Friedel-Crafts-artige elektrophile Substitution II. Direkte Aurierung O O+ O 1 Mol-% AuCl3 MeCN, 20°C [Au] H [Au] oder O + + O O O Protodesaurierung - [Au] [Au] O + + H H O + + H O C) Alkene Reaktion von Bis(m-oxo)-Gold(III)-Komplexe mit Norbornen 2+ R N O Au N 2 [PF6]- Au N O R = z.B. Me, iPr N R O MeCN, H2O, RT • Gold-Komplex katalysiert die Addition von Wasser an ein Alken • liefert das Epoxid in einer stöchiometrischen Reaktion C) Alkene Erstes und bislang einziges isoliertes metallorganisches Analogon einer Zwischenstufe einer Gold-katalysierten Reaktion (Cinellu et al.) + R N O Au [PF6]- N Auraoxetan Konnte vom Alken-Komplex abgetrennt werden und mittels Röntgenkristallstrukturanalyse zweifelsfrei charakterisiert werden Nucleophile Additionen an Allene Cycloisomerisierung von Allenylketonen • 2000: Hashmi et. al. • Wheland Zwischenstufe • Aromatisierung • Protodesaurierung Die Oxidationsstufe des Goldes beeinflusst die Konstitution Nucleophile Addition an Alkinen Bildung von Furanen aus Alkinylketonen • Katalysator koordiniert an Dreifachbindung • Bildung eines Furanlykations • Addition von Methanol Bildung von Naphthalinen • Bildung einer Pyrylium Zwischenstufe • [4+2] Cycloaddition mit Alkinen oder Alkenen und Ringöffung • Mechanismus wurde durch Rechnung bestätigt Cyclisierung von Alkinphenolen od. -anilinen • Bildung aus ortho substituierten Aromaten • X=O: AuCl3 • X=NH: Na[AuCl4]∙H2O Ringerweiterungsreaktionen Epoxidringerweiterung • Liefert interessantes Substitutionsmuster der Furane • R1: -Me, -Et-OH • R2: -Pr-OH, -Bu-OH, -Ph • Nuc. Angriff des Epoxids schneller als der der Hydroxygruppe Bildung von Cyclobutanon aus Cyclopropanol Gold Katalyse Nukleophile Additionen an π -Systeme Gliederung: 1. Reaktionen von ambidenten nukleophilen Gruppen in Propargyl – Position 1.1 Reaktionen über Vinylcarbenoide 1.2 Reaktionen über Allenylester 1.3 Reaktionen über Vinylgold-Spezies 1. Gold-katalysierte Reaktionen ambidenter Gruppen in Propargyl-Position Zwei unterschiedliche Reaktionswege: 5-exo-dig – und 6-endo-dig Modus Reaktionsmodi 5-exo-dig 6-endo-dig Allen Vinylcarbenoid Reaktionen über Vinylcarbenoide O O [Au] O O O + O Ph Alkenylester 26% 2 Diastereomere 63% Reaktionen über Vinylcarbenoide Anwendung - Synthese von Caren – Terpenoiden durch Abfangen der Vinylcarbenoide durch Doppelbindung in passender Entfernung O O O [Au] O O O 95% Vinylcarbenoid • gute Diastereoselektivität • nur marginale Mengen an isomeren Allenylacetats Reaktionen über Allenylester Au+ Nazarov Cyclisierung Mechanismus der Reaktion von Allenylestern O O O O [Au] + C Pent Pent O O O O O [Au] + [Au] C H + H Pent Pent Pent H2O 1,2 Hydridshift Hydrolyse - HOAc H3O+ O O O O [Au] + H O - [Au]+ O [Au] + C Pent Pent Pent 1.3 Reaktionen über Vinylgold - Spezies - 5-exo-dig und 6-endo-dig Reaktionen laufen über Vinylgold – Spezies - Befindet sich eine NH-Gruppe in Propargylposition, kann ein Proton auf dieser Stufe eliminiert und der Goldkatalysator durch Protodemetallierung freigesetzt werden 1.3 Reaktionen über Vinylgold - Spezies N-Propargylcarboxamid Oxazol R R R + O O N H C N [Au] [Au] R langsam O N schnell H O - [Au] N