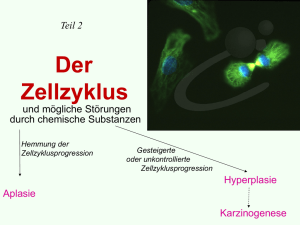
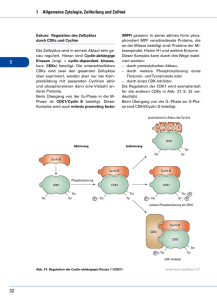
Die Funktion und Regulation von Cyclin A in den frühen Kernzyklen
Werbung

Die Funktion und Regulation von Cyclin A in den frühen Kernzyklen von Drosophila melanogaster Diese Diplomarbeit erstellt an der Universität zu Köln am Institut für Genetik wurde dem Prüfungsausschuß der Universität Bonn vorgelegt von Christine Schulze aus Reutlingen Dezember 1998 meinem Ehemann Danksagung Die vorliegende Arbeit wurde vom 15. März 1998 bis zum 15. Dezember 1998 am Institut für Genetik an der Universität Köln in der Abteilung von Frau Prof. Dr. M.Leptin unter Anleitung von Dr. Frank Sprenger angefertigt. Mein Dank gilt Dr. Sprenger für die Bereitstellung des Arbeitsplatzes und der finanziellen Mittel, die die Durchführung der Arbeit ermöglichten. Weiterhin danke ich Dr. Sprenger für die jederzeit vorhandene Bereitschaft meine Arbeit wissenschaftlich zu fördern und mit kritischem Engagement zu begleiten. Ausserdem stellte er mir freundlicherweise 3 Abbildungen (Abb. 20, 21 und 22) zur Verfügung, die Eingang in die vorliegende Diplomarbeit fanden. Der Abteilung verdanke ich darüberhinaus viele wertvolle Anregungen in experimentellen Details. Insbesondere möchte ich hier den Beitrag aller Mitglieder der Arbeitsgruppe von Dr. Sprenger, Ruth Grosskortenhaus, Edan Foley, Axel Dienemann und Hayati Özdem, herausstellen. Alle zusammen sorgten über die gesamte Zeit dafür, daß die Arbeit in einer entspannten und aufmunternden Atmosphäre durchgeführt werden konnte. Die in der vorliegenden Diplomarbeit vorgestellten Resultate sind das Ergebnis meiner eigenen Arbeit. Nicht eigene Daten sind als solche an dortiger Stelle entsprechend gekennzeichnet. Christine Schulze Inhaltsverzeichnis 1. Einleitung 1.1. Der Zellzyklus 1.2. Cycline und Cdks 1.2.1.Klassifikation 1.2.2. Regulation 1.2.2.1.Aktivierung/Inhibition durch Phosphorylierung 1.2.2.2. Kontrolle duch Degradation von Cyclinen 1.3. Die frühe Embryogenese bei Drosophila 1.3.1. Die Funktion des Cyclin A während der frühen Kernzyklen 1 1 2 2 2 2 2 3 4 2. Material und Methoden 2.1. Chemikalien und Enzyme 2.2. Methoden 6 6 9 3. Ergebnisse 3.1. Analyse der Stabilität von Cyclin A 3.1.1. Das mit HA-Cyclin A und HA-Cyclin A∆169 assoziierte Cdk1 (cdc2) zeigt im Kinase-Assay Aktivität 3.1.2. In vitro Analyse zur Stabilität von Cyclin A 3.1.3. In vitro Analyse zur Stabilität von Cyclin B 3.1.4. Einfluß von Proteasomeninhibitoren auf den Cyclin-Abbau 3.1.5. Caspase-Inhibitoren 3.1.6. Der Einfluß des Alters bzw. des Entwicklungsstadiums der Embryonen auf die Stabilität von Cyclin A 3.1.7. Die Stabilität von endogenem Cyclin A und Cyclin B 3.2. Keimbahntransformation der HA-Cyclin A und HA-Cyclin A∆169 Gene 3.2.1. Klonierung der zu injizierenden HA-Cyclin A und HA-Cyclin A∆169 Konstrukte zur Erzeugung transgener Fliegen 3.2.2. Erzeugung transgener Fliegen 3.2.3. Nachweis von HA-Cyclin A und HA-Cyclin A∆169 in transgenen Fliegen 3.3. Injektion von RNA in Drosophila Embryonen 3.3.1. Testung der Transkriptionsbedingungen 3.3.2. Einfluß der in vivo translatierten Cyclin A Konstrukte auf den Zellzyklus 15 15 18 20 25 26 30 32 34 37 38 39 40 41 41 43 4. Diskussion 4.1. In vitro Untersuchungen zur Stabilität von Cyclin A 4.2. Die Funktion der Stabilität von Cyclin A in den frühen Kernzyklen 51 51 53 5. Zusammenfassung 56 6. Literatur 57 1 1. Einleitung 1.1. Der Zellzyklus Am Anfang der Entwicklung eines jeden mehrzelligen Organsimuses steht eine befruchtete Eizelle. Nur durch viele aufeinanderfolgende Teilungen und Differenzierungsschritte dieser Eizelle ist die Entstehung eines Mehrzellers erst möglich. Diese Teilungs- und Differenzierungsereignisse werden aufwendig reguliert. Insbesondere besteht eine enge Verknüpfung zwischen Zellteilung einerseits und Zellwachstum sowie DNA Replikation andererseits. Ein typischer Zellzyklus besteht aus 4 Phasen die als M-, G1- S- und G2-Phasen benannt wurden. Im einzelnen entspricht die M Phase der Mitose, die G1 Phase charakterisiert das Intervall zwischen Zellteilung und einsetzender DNA Replikation in der die metabolisch aktive Zelle kontinuierlich wächst. Die S Phase beschreibt die Duplikation der DNA, woraufhin die Zelle in der abschließenden G2 Phase weiterhin an Größe gewinnt und die zur endgültigen Zellteilung benötigten Proteine synthetisiert. Die Dauer und der Übergang zwischen den einzelnen Phasen wird durch die zyklische Aktivität einer Klasse von Proteinkinasen, der Cdks (cyclin-dependent kinases) reguliert. Diese Proteinkinasen brauchen für ihre Aktivität die Bindung einer regulatorischen Untereinheit, der Cycline. Infolge der Bindung an unterschiedliche Cycline ergeben sich verschiedene Kombinationen aus Cdks und Cyclinen, welche den zeitlichen Verlauf der aufeinanderfolgenden Zellzyklusstadien kontrollieren (Nurse, 1994; Evans, 1983; Lohka, et al., 1988). Konkret wird die Aktivität der Cdks daher durch die Assoziation mit unterschiedlichen Cyclinen, aber darüberhinaus auch durch aktivierende und inhibitorische Phosphorylierungen und die mögliche Bindung von Cdk Inhibitoren gesteuert. Zusätzlich etablieren sich während des Ablaufs des Zellzyklus biochemische Rückkopplungsschleifen, sog. Checkpoints. Hierbei wird z.B. die Intaktheit der DNA „gemessen“ und bei Schäden Proteine aktiviert die den Zellzyklusverlauf stoppen bis die Schäden behoben sind. Damit steht der Zelle eine übergeordnete Kontrolle zur Verfügung, vor deren Hintergrund die zyklische Aktivität der Cdk/Cyclin-Komplexe ihre regulatorische Funktion wahrnimmt. Die wesentlichen Experimente zur Analyse des Zellzyklus wurden im System der Hefe und mit Xenopus Eiern durchgeführt. Wie sich dann zeigte konnten die zentralen Befunde fast vollständig auf andere Spezies, darunter Drosophila und Mensch, übertragen werden. 2 1.2. Cycline und Cdks 1.2.1.Klassifikation Die periodische Bildung, Aktivierung und Dissoziation von Cyclinen und Cyclin-abhängigen Proteinkinasen steuern den Zellzyklus. Grundsätzlich ist die jeweilige Zellzyklusphase charakterisiert durch die Art des Cdk-Cyclin Dimers. Dabei sind zwei Hauptklassen der Cycline bekannt. Die mitotischen Cycline, Cyclin A und Cyclin B, assoziieren während der G2-Phase in Säugerzellen mit Cdk1 und sind verantwortlich für den Einstieg in die Mitose. Die für die G1-Phase charakteristischen Cycline, Cyclin E, Cyclin D, sind verantwortlich für den Eintritt in die S-Phase. Hierbei assoziiert Cyclin E mit Cdk2 und Cyclin D mit der Cdk4 und der Cdk6 Kinase. Die Progression durch die S-Phase erfordert die Anwesenheit eines funktionell aktiven Cdk2/Cyclin A Komplexes. Zum Eintritt in die Mitose wird Cyclin A erneut benötigt zur Interaktion mit Cdk1. Damit fällt dem Cyclin A als einziges Cyclin eine duale Funktion zu, da es sowohl in der S-Phase sowie für den Übergang von G2 nach M benötigt wird (Girard, 1991; Pagano, 1992). 1.2.2. Regulation 1.2.2.1. Aktivierung/Inhibition durch Phosphorylierungen Die Aktivität der Cyclin/Cdk Dimere wird durch spezifische Phosphorylierungen an bestimmten Tyrosin und Threonin Resten reguliert. Nach der Bildung des Cyclin-Cdk Komplexes erfolgt eine Phosphorylierung eines konservierten Threonin-Restes an Position 161 (T161) der Cdks, die für eine vollständige Aktivierung von Cdks notwendig ist. Diese Phosphorylierung erfolgt in höheren Eukaryonten durch ein Heterotrimer Komplex, der CAK (Cdk Activating Kinase) genannt wird (Draetta, 1997; Larochelle et al. 1998). Weiterhin können Cdks durch inhibitorische Phosphorylierungen am Threonin 14 (T14) und am Tyrosin 15 (Y15) reguliert werden. Hierfür verantwortlich sind die Kinasen Wee und die Myt-like Kinase (Dunphy, 1994). Eine aktive Cdk-Kinase benötigt ein gebundenes Cyclin, die Phosphorylierung des T161 und die Abwesenheit der inhibierenden Phosphorylierung am T14 und Y15. 1.2.2.2. Kontrolle durch Degradation von Cyclinen Nach der Regulation durch Phosphorylierung, wird die Aktivität der Cdks im Zellzyklus durch die Fluktuation der Cyclin-Untereinheiten reguliert. Hierfür ist im wesentlichen der Cyclin-Abbau während der Mitose von Bedeutung. An diesem Mechanismus ist ein großer Proteinkomplex, der „Anaphase-Promoting Complex“ (APC) oder auch „Cyclosom“ genannt, beteiligt (Sudakin, et al., 1995; King, et al., 1995). Dieser Komplex ist für den Abbau von Anaphase-Inhibitoren und den mitotischen Cyclinen verantworlich und leitet dadurch die Trennung der Chromosomen und den Ausstieg aus der Mitose ein. Der Vorgang der Proteolyse steuert den Verlauf des Zellzykluses nicht allein durch den Abbau Cdk 3 regulierender Proteine, sondern hat auch direkten Einfluß auf die Dynamik der Chromosomen und des Spindelapparates. Der APC wird während der M-Phase durch Cdk1 aktiviert, und induziert dadurch den Abbau des Cyclins mit der die Kinase selbst assoziiert ist. Dieser Abbau der mitotischen Cycline während der M-Phase ermöglicht der Zelle den Austritt aus der Mitose. Der APC ist während der gesamten G1Phase aktiv und verhindert ein Akkumulieren der mitotischen Cycline was einen vorzeitigen Einstieg in die Mitose und S-Phase zur Folge hätte. In der S-Phase wird der APC wieder inaktiviert, dadurch können M-Phase Cycline wieder akkumuliert werden, die für einen erfolgreichen G2-M Übergang benötigt werden (Evans, 1983; Murray, and Kirschner, 1989). Der APC vermittelt die Polyubiqutinierung von Proteinen die anschließend durch das Proteasom abgebaut werden. Für diesen Vorgang wird zunächst ein Ubiquitin-Aktivierungs -enzym (E1) benötigt. Das Ubiquitin wird an seinem COOH-Ende durch eine Thioester-Bindung mit dem Ubiquitin-Aktivierungs Enzym E1 aktiviert. Dieses Ubiquitin wird dann auf ein Ubiquitin-conjugating Enzym (E2 oder UBC genannt) übertragen (Hershko, 1983). Vom E2 gelangt Ubiquitin schließlich auf ein Lysin-Rest eines Zielproteins (Scheffner, 1995). Dieser Prozeß erfolgt entweder direkt oder mit Hilfe der UbiquitinProtein Ligase (E3) (Hershko, 1983). In diesem Fall agiert APC als E3 Enzym (Bartel B., 1990). Das APC ubiqutiniert nur solche Proteine die eine Erkennungssequenz tragen. Diese Erkennungssequenz wird als destruction box bezeichnet. In der Nähe dieser destruction box befinden sich einige Lysine, die als Ziel der Polyubiquitinierung dienen. Die ubiqutinierten Lysine vermitteln schließlich die Substraterkennung durch das Proteasom. Die 26S Untereinheit des Proteasoms ist verantwortlich für den Abbau von Proteinen mit einer Polyubiquitinkette (Peters, 1994; Hochstrasser, 1995). Die Rate und Spezifität des Ubiquitinvermittelten Proteinabbaus wird zusätzlich zur substratspezifischen Ubiquitinierung auch durch die Deubiquitinierung gewährleistet. Die Deubiquitinierung wird durch eine bisher noch wenig bekannte Familie der deubiquitinierenden Enzyme (UBPs) katalysiert (Papa, 1993; Huang, 1995; Zhu, 1996). 1.3. Die frühe Embryogenese bei Drosophila Bisher wurde der „generelle“ Zellzyklus, wie er zum Beispiel in humanen Zellen in Gewebekultur stattfindet, beschrieben. Dieser „generelle“ Zyklus besteht aus den 4 Phasen G1, S, G2 und M. Während der Entwicklung von multizellulären Organsimen findet man jedoch zusätzlich „spezielle“ Zellzyklen. So bestehen zum Beispiel die ersten Zellzyklen in Drosophila nur aus S- und M-Phase, Gap-Phasen gibt es nicht. Die Analyse der Zellzykluskontrolle während der Entwicklung eines Organismus sollte daher berücksichtigen, daß nicht nur die Regulation der „generellen“-Zellzyklen, sondern auch die Besonderheiten der speziellen Zellzyklen mit einbezogen werden. 4 Während der Drosophilaentwicklung werden verschiedene Arten von Zellzyklen, die mitunter gleichzeitig in einem Embryo ablaufen können, beobachtet. Nach den meiotischen Teilungen, die durch die Eiablage ausgelöst werden, teilen sich die Kerne 13 mal synchron im Embryo. Diese Zyklen bestehen nur aus S- und M-Phasen. Die Kontrolle dieser Zyklen soll im Rahmen dieser Diplomarbeit genauer untersucht werden. In der Interphase 14 findet die Zellularisierung statt und alle Zellen gehen nach der DNA-Replikation in eine längere G2-Phase. Der Eintritt in die Mitose wird in diesem und den darauffolgenden nächsten drei Zellzyklen durch die String-Phosphatase reguliert (Edgar, and O’Farrell, 1989; Edgar, and O’Farrell, 1990). Nach diesen String-kontrollierten Zyklen gehen die meisten Zellen in die erste G1-Phase. Für den Übergang in die nächste S-Phase ist die Expression von Cyclin E limitierend (Duronio, et al., 1995; Knoblich, et al., 1994). Auch dieser G1/S-Übergang ist zeitlich und räumlich reguliert. Die Signale, die hierfür verantwortlich sind, sind jedoch nicht bekannt. Die meisten Zellen des Embryos gehen anschließend durch Endozyklen, in denen S-Phase und Gap-Phasen einander ablösen. Da diese Zellen keine Mitose ausführen, werden sie polyploid (Duronio, and O’Farrell, 1994; Edgar, and O’Farrell, 1989). 1.3.1. Die Funktion des Cyclin A während der frühen Kernzyklen Das Cyclin A wird während des zellulären Blastoderm-Stadiums zusammen mit Cyclin B für den G2/M-Übergang benötigt (Knoblich, and Lehner, 1993). In Xenopus wurde gezeigt, daß das Cyclin A während der Mitose den Abbau des Zellkernes induziert und für die Kondensation der Chromosomen verantwortlich ist (Roy, 1991; Swenson, 1986). In Drosophila haben genetische Studien von DmcycA gezeigt, daß sich die Funktionen von Cyclin A in der Mitose mit denen von Cyclin B überschneiden (Knoblich, 1993). In Zellen von Vertebraten bildet Cyclin A sowohl mit Cdk1 wie auch mit Cdk2 Komplexe. Dabei wurde die Funktion von Cyclin A in der S-Phase durch den Cyclin A/Cdk2 Komplex und während der G2-Phase durch den Cyclin A/Cdk1 Komplex bestimmt (Girard, 1991; Pagano, 1992; Rosenblatt, 1992; Resnitzky, 1995; Tsai, 1991; Hamaguchi, 1992). In Drosophila assoziiert Cyclin A nur mit Cdk1 (cdc2) und wird sowohl in der S-Phase wie auch in der G2-Phase als Cyclin A/Cdk1 Komplex funktionell (Knoblich, 1994; Sauer, 1995). Die ersten 13 Proliferationszyklen des Drosophila-Embryos sind synchrone Kernteilungen. Mit einer Zykluszeit von nur 8,6 Minuten sind diese Zyklen die schnellsten uns bekannten Zellzyklen. Dadurch ist es möglich in kurzer Zeit das Genom zu multiplizieren und zahlreiche Zellen zu bilden, die dann im Rahmen der Musterbildung spezialisiert werden können. Wie diese Zellzyklen reguliert werden und was die Verlängerung der Interphase während der Zyklen 8-13 bewirkt, ist im einzelnen noch nicht bekannt. Die ersten 13 Zyklen werden von maternalen Komponenten gesteuert und sind deshalb genetisch schwer untersuchbar. In den Zyklen 8-13 findet eine kontinuierliche Translation der maternalen Cyclin mRNAs statt. In Folge des periodischen Abbaus während des Metaphasen/Anaphasen- 5 Übergangs fluktuieren die Cycline in diesen Zyklen. Nach dem mitotischen Abbau ist zu Beginn der Interphase die Cdk1-Kinaseaktivität niedrig. Zum Eintritt in die nächste Mitose ist die erneute Akkumulierung der Cycline notwendig (Edgar, et al., 1994). In den ersten sieben Zyklen findet jedoch keine Fluktuation von Cyclinen statt und die Zellzykluslänge wird durch Verminderung des maternalen Beitrages der Cycline nicht verändert. Die Cdk1-Kinaseaktivität zeigt ebenfalls keine nachweisbaren Schwankungen, das heißt die Kinase ist in den ersten Zyklen ständig aktiv, auch in der Interphase (Edgar, et al., 1994). Wie es möglich ist, in der Gegenwart von hoher Cdk1Kinaseaktivität aus der Mitose in die Interphase zu gelangen und wie diese Interphase aufrecht erhalten werden kann bis die DNA vollständig repliziert ist, konnte bisher noch nicht geklärt werden. Für den Austritt aus der Mitose ist normalerweise die Inaktivierung von Cdk1 durch Cyclin-Abbau notwendig (Murray, et al., 1989). Ein Abbau von Cyclinen kann in den ersten Mitosen jedoch nicht nachgewiesen werden. Erst ab Zyklus 8 wird Cyclin B erkennbar abgebaut, wobei die Menge an Cyclin B Abbau kontinuierlich ansteigt, so daß während der Mitose 13 fast sämtliches Cyclin B verschwindet. Der Abbau von Cyclinen während der Mitose geschieht durch Ubiquitin-abhängige Proteolyse (Murray, 1995). Für diesen Prozeß muß die destruction box im Substrat enthalten sein (Glotzer, et al., 1991). Cycline, denen die destruction box fehlt, oder die eine Mutation in der destruction box aufweisen, können in der Mitose nicht abgebaut werden und verhindern den Austritt aus der Mitose (Holloway, et al., 1993; Luca, et al., 1991; Sigrist, et al., 1995). Injiziert man stabile Cyclin B Proteine in Drosophila Embryonen, führt dies zu einem Mitoseblock. Dies Beobachtung zeigt, daß für den normalen Austritt aus der Mitose der Abbau von Cyclin B selbst in den Zyklen notwendig ist, in denen kein Abbau nachweisbar ist (Su, et al. 1998). Wie in der folgenden Interphase trotz hoher Cdk Aktivität ein vorzeitige Mitose verhindert werden kann, ist die zentrale Frage dieser Arbeit. Als Ansatzpunkt liegt hierbei eine Beobachtung zugrunde, die zeigt, daß Cyclin A und Cyclin B verschiedene Halbwertszeiten besitzen. Die Halbwertszeiten von Cyclin A und B wurde in Embryonen getestet die durch Zugabe von Cyclohexamid in der Interphase arretierten. Es zeigte sich in diesem Versuch, daß Cyclin B während der ersten 8 Kernteilungen stabil ist, während Cyclin A abgebaut wurde. In den Zyklen 10-13 ist Cyclin A in der Interphase stabil (Edgar, et al., 1994). In den frühen Kernzyklen von Drosophila ist Cyclin A zwar vorhanden, aber nur wenig Cyclin A ist mit Cdk1 assoziiert (Sprenger, and O’Farrell, unveröffentlicht). Welcher Mechanismus für die Instabilität von Cyclin A während der Zyklen 2-9 verantwortlich sind, soll im Rahmen dieser Diplomarbeit untersucht werden. Hierfür sollte zunächst ein in vitro System etabliert werden, das es ermöglicht, die Mechanismen, die für die unterschiedliche Stabilität verantwortlich sind, zu untersuchen. Die daraus resultierenden Ergebnisse sollten anschließend durch in vivo Expression von unterschiedlichen Cyclin A Konstrukten untersucht werden. 6 2. Material und Methoden 2.1. Chemikalien und Enzyme 2.1.1.Chemikalien Agar Gibco BRL. Eggenstein Agarose Gibco BRL. Eggenstein Diethylpyrocarbonat (DTT) Sigma. St. Louis, USA Ethidiumbromid Sigma. St. Louis, USA Glycin Sigma. St. Louis, USA Isopropanol Roth. Karlsruhe Klorix Dan Chemie. Hamburg m7G(5’)ppp(5’)G Pharmacia. Freiburg Natriumdodecylsulfat (SDS) Serva. Heidelberg Nukleotide: dATP, dCTP, dGTP, dTTP Pharmacia. Freiburg rATP, rCTP, rGTP, rUTP Phenol Roth. Karlsruhe Triton X-100 Serva. Heidelberg Tween 20 Sigma. St. Louis, USA Voltalev Öl PCTFE, 3S und 10S Atochem, Pierre-Benite, Frankreich Alle weiteren, nicht aufgeführten Chemikalien wurden von der Firma Merck, Darmstadt bezogen. 2.1.2. Radiochemikalien 35 S-Methionin 15 mCi/ml Amersham, Braunschweig 2.1.3. Enzyme CIP Boehringer Mannheim Klenow-Fragment aus E.coli Boehringer Mannheim Restriktionsenzyme: Asp718, BamHI, BamHIII, Bgl I, Boehringer Mannheim bzw. EcoR , EcoRIII, HindI, HindIII, New England Biolabs, USA bzw. Not I, Nco I, Sac I, Xba I, Pharmacia, Freiburg RNase H Sigma, St. Louis, USA RNase Inhibitor Boehringer Mannheim T4 DNA -Ligase Boehringer Mannheim SP6 Polymerase Boehringer Mannheim 2.1.4. Inhibitoren MG-132 Calbiochem Novabiochem GmbH Calpain Inhibitor I Calbiochem Novabiochem GmbH Ac-YVAD-CHO Biomol GmbH, Hamburg DEVD-CHO Biomol GmbH, Hamburg 2.1.5. DNA Basenpaar Standard Lambda DNA-BstEII Digest New England Biolabs, USA 2.1.6. Antikörper Anti-HA aus Ratte Boehringer Mannheim Anti-Ratte-HRP (horse-radishBoehringer Mannheim peroxidase) 2.1.7. Drosophila melanogaster Oregon R Fliegenembryonen wurden benutzt um Embryonenextrakt herzustellen. White--Fliegen wurden zur erzeugung transgener Fliegen benötigt. Die Embryonen von white+-Fliegen wurden zur RNA Injektion verwendet. 7 2.1.8. Escherichia coli Transformationen wurden durchgeführt mit DH10B, Genotyp; F’, mcrAD- (mrr hsdRMS-mcrBX), j80dlacZDM15, DlacX74, deoR, recA1, araD139, D(ara, leu) 7694, galK, l- rspL, endA1, nupG. 2.1.9. Kaninchen Retikulozyten Lysat Rabbit reticulocyte lysate Promega 2.1.10. Filme Hyperfilm ECL Kodak Biomax MR Amersham, Braunschweig Kodak, USA 2.1.11. Lösungen und Puffer Embryo-Homogenisationspuffer 10mM Tris pH 7,5 80mM K-β-Glycerophosphat pH 7,3 20mM EGTA 15mM MgCl2 10% Glycerol Kurz vor Gebrauch, Zugabe von: 1mM Benzamidin 2mM Na2VO4 1mM DTT Unmittelbar vor Gebrauch, Zugabe von: 25µg/ml Aprotinin 25µg/ml Leupeptin 0,5mM PMSF Laemmli Proben Puffer (4X) 8% SDS 400mM DTT 240mM Tris pH 6,8 0,004% BPB 40% Glycerol Ligationsmix I 100mM MgCl2 100mM Tris/HCl pH 8,0 100mM DTT II 1mg/ml BSA in ddH2O III 500mM Tris/HCl pH 8,0 50mM DTT 10mM ATP Alle drei Lösungen werden im gleichen Verhältnis vor Gebrauch gemischt. 8 DNA Mini-Präparations Puffer P1: Resuspensionspuffer 100µg RNase A 50mM Tris/HCl 10mM EDTA pH 8,0 P2: Lysispuffer 200mM NaOH 1% SDS P3: Neutralisatiospuffer 3M Kac pH 5,5 Bindungssuspension 1 : 1 Suspension von Diatomecous earth (Sigma D-5384) in 7M Guanidinium/HCl Waschlösung 100mM NaCl 10mM Tris pH 7,0 2,5mM EDTA 50% Ethanol 10X Ponceau S 2g Ponceau S 30g Trichloressigsäure 30g Sulfosalicylsäure 10X PBS 1,47M NaCl 26,8mM KCl 80mM Na2HPO4 17,6mM KH2PO4 mit HCl auf einen pH-Wert von 7,4 einstellen 10X PBT 0,1% TWEEN 20 in PBS 10X Laufpuffer für SDS-PAGE 0,25M Tris-base 1,9M Glycine 1% SDS 1L 4X Trenngelpuffer 181,7g Tris base (1,5M) 4ml 10% SDS Mit HCl einen pH-Wert von 8,8 einstellen. 500ml 4X Sammelgelpuffer 30,3g Tris base (0,5M) 20ml 10% SDS Mit HCl einen pH-Wert von 6,8 einstellen. 1l Transferpuffer für Western-Blots 5,82g Tris (48mM) 2,93g Glycine (39mM) 3,75ml 10% SDS 200ml Methanol 9 TE 10mM Tris/HCl pH 8,0 1mM EDTA pH 8,0 1l 50X TAE 2,42g Tris base (2M) 57,1ml Eisessig 500mM EDTA pH 8,0 Terrific broth A 12g Bactotrypton 24g Bacto-yeast-extract 4ml Glycerol auf 900ml mit ddH2O auffüllen. B 2,31g KH2PO4 12,54g K2HPO4 auf 90ml mit ddH2O auffüllen. Lösung A und B werden vor Gebrauch gemischt. 1l LB-Medium 10g Bacto-Trypton 5g Bacto Yeast Extract 10g NaCl Mit NaOH auf pH 7,0 eingestellt. 2.2. Methoden 2.2.1. Herstellung von Embryonenextrakt und in vitro Stabilitätsversuche 2.2.1.1. Sammeln von Embryonen In zylindrischen Fliegenkäfigen („Legekäfigen“) aus Kunststoff wurden ca. 400-500 Fliegen des jeweils benötigten Genotyps gehalten. Zur Herstellung von Embryoextrakt wurden Wildtyp-Fliegen verwendet. Die obere Öffnung des Kunststoffzylinders war mit einem feinmaschigen Stahlnetzchen bespannt, auf der Unterseite wurden Apfelsaft-Agarplatten passender Größe mit Hilfe eines Klebestreifens befestigt. Die Mitte der Apfelsaft-Agarplatte wurde mit etwas Bäckerhefe versehen. Das erste Tagesgelege in einem Legekäfig wurde verworfen, da die abgelegten Eier in Bezug auf ihr Alter noch nicht „synchronisiert“ waren. Bei den nachfolgenden Gelegen, die gesammelt wurden, waren die Eier zum allergrößten Teil frisch befruchtet, was die Voraussage der Entwicklung möglich macht. Die gelegten Eier wurden nach gewünschten Zeitintervallen vorsichtig mit einem Spatel von der Platte gekratzt. Diese Eier wurden in einem Eppendorf-Schraubgefäß gegeben und in Flüssigstickstoff schock gefroren. Diese Eier konnten nun bei -70C° bis zum Gebrauch aufbewahrt werden, oder sofort zu Embryonenextrakt weiterverarbeitet werden. 2.2.1.2.Herstellung von Embryonenextrakt Wildtyp-Eier wurden wie oben beschrieben gesammelt und schock gefroren. Eine Probe von ca. 0,1g Embryonen wurden in 1ml Homogenisationspuffer mit Hilfe eines Homogenisators auf Eis homogenisiert. Das Homogenisat wurde anschließend in einem Eppendorf-Reaktionsgefäß 15 Min. bei 4C° und 14 000rpm zentrifugiert. Die mittlere Phase wurde in ein neues EppendorfReaktionsgefäß gegeben und erneut unter gleichen Bedingungen 15 Min. zentrifugiert. Wiederum wurde die mittlere Phase entnommen, in 50µl Aliquots schock gefroren und anschließend bei -70C° gelagert. 10 2.2.1.3. In vitro Transkription Ein 20µl Ansatz bestand aus: 4µl 5 x Transkriptionspuffer (200mM Tris/HCl pH 7,5; 30mM MgCl 2; 10mM Spermidin) 2µl DTT (0,1M) 1µl RNasin (20U/µl) 4µl 5 x rNTPs (ATP, UTP, CTP und GTP je 2,5mM) 5U SP6 Polymerase Der Ansatz wurde für zwei Stunden bei 37C° inkubiert und anschließend bei -20C° gelagert. 2.2.1.4. In vitro Translation Ein 50µl Ansatz bestand aus: 35µl Kaninchen Reticulozyten-Lysat 1µl 1mM Aminosäuremix 4µl RNasin (40U/µl) 4µl RNA (1µg/µl) 6µl ddH 2O Der Ansatz wurde für zwei Stunden bei 30C° inkubiert. Für eine Translationskontrolle, wurde eine 2µl Probe entnommen und mit 5µl Laemmli gemischt. Anschließend wurde die Probe für 5 Min. bei 95C° erhitzt und auf einem SDS-Polyacrylamidgel aufgetrennt. Mit Hilfe des Western-Blots wurde die Probe auf ihre Richtigkeit hin überprüft. 2.2.1.5. Embryonenextrakt-Versuche Für einen Stabilitätsversuch ohne Inhibitor: 25µl Embryonenextrakt 10µl in vitro translatiertes Protein 2mM ATP 15mM MgCl2 auf 40µl mit ddH 2O aufgefüllt. Wurde dem Ansatz ein Inhibitor oder Ubiquitin zugesetzt, verringerte sich die Zugabe des destilierten Wassers. Der Kontrolle, ohne Inhibitor oder Ubiquitin, wurde immer die entsprechende Menge des Lösungsmittels z.B. DMSO zugegeben. Für den 0 Min. Wert wurde dem Ansatz, vor Inkubation bei 25C°, 7µl entnommen und mit 5µl Laemmli gemischt. Weitere 7µl Probe wurden nach 15 Min., 45 Min. und 70 Min. entnommen. Allen Proben wurde 5µl Laemmli zugegeben und 5 Min auf 95C° erhitzt. 2.2.1.6. Western Blot Proteinextrakt wurde auf ein 10%iges SDS-Polyacrylamidgel aufgetragen. Die Elektrophorese wurde bei konstanter Stromstärke durchgeführt. Die Proteine wurden anschließend elektrophoretisch mittels „Western Blotting“ auf Nitrozellulose bei konstanter Spannung (5,5mA/cm_) in einer Multiblot Kammer (Pharmacia) übertragen. Die Nitrozellulosefilter wurden eine Stunde in PMT (5% Magermilchpulver in PBT) bei 4C° geblockt und über Nacht mit dem ersten Antikörper in PMT ebenfalls bei 4C° inkubiert. Die Nitrozellulose wurde dreimal 20 Minuten mit PBT gewaschen und für eine Stunde bei 4C° mit Peroxidase-konjugierten zweiten Antikörper inkubiert. Es wurde dreimal 5 Minuten mit PBT gewaschen. Anschließend wurde die Nitrozellulose mit den ECLSystem (Amersham) nach Herstellerangaben behandelt und die Signale durch Autoradiographie mit einem Röntgenfilm nachgewiesen. 2.2.2. Präparation von DNA 2.2.2.1. Restriktionsendonukleaseverdau Der Verdau von DNA mit Restriktionsendonukleasen (Typ II) wurde unter den jeweils geeigneten Salz- und Temperaturbedingungen durchgeführt (Herstellerangaben bzw. Maniatis et al., 1982). Die Menge an Enzym bzw. die Inkubationsdauer wurde der zu verdauenden DNA-Menge angepaßt. 2.2.2.2. Elektrophoretische Auftrennung von DNA-Fragmenten in Agarosegelen Restriktionsverdaute DNA wurde in horizontalen Agarosegelen in Gegenwart von 0,4µg/ml Ethidiumbromid aufgetrennt. Die Konzentration der Gele betrug in der Regel 1% und wurden 11 entsprechend der Größe der DNA-Fragmente gewählt. Für präparative Gele wurde eine Low Melting Agarose verwendet, die eine DNA-Extraktion mit AgarACE (nach der Vorschrift von Promega) ermöglichte. Die in 1 x TAE suspendierte Agarose wurde unter Kochen gelöst. Nach Abkühlung auf etwa 50°C wurde Ethidiumbromidlösung zugegeben und die Lösung in eine abgeklebte Gelwanne mit eingehängtem Kamm gegossen. Nach Erstarren der Agarose wurde das Gel in eine Elektrophoresekammer gelegt, mit Elektrophoresepuffer (1xTAE) überschichtet und der Kamm entfernt. Die Proben wurden mit 0,2 Vol DNA-Ladepuffer versetzt und in die Geltaschen geladen. Die Auftrennung der DNA-Fragmente erfolgte bei Spannungen zwischen 50V und 100V. Nach Beendigung der Elektrophorese wurde das Gel auf eine UV-Lampe (366nm) gelegt, um die DNA-Fragmente sichtbar zu machen und zu fotografieren. Bei einem präparativen Gel wurde eine schwächere UV-Handlampe verwendet und die DNA-Fragmente mit Hilfe eines Skalpells aus dem Gel herausgeschnitten. Als Größenstandard wurde die λ-BstE II DNA Leiter verwendet. 2.2.2.3. DNA-Extraktion aus dem Gel Die DNA wurde mit Hilfe von AgarACE aus der Low Melting Agarose nach der Methode der Firma Promega extrahiert. 2.2.2.4. Dephosphorylierung von DNA-Enden Geschnittene und evtl. aus dem Gel extrahierte Vektor-DNA wurde in 44µl ddH2O aufgenommen. Nach Zugabe von 5µl Dephosphorylationspuffer (Boehringer Mannheim) und 1µl CIP (Calf Intestinal Phosphatase, 50U/µl; Boehringer Mannheim), wurde der Ansatz für 15 Minuten bei 37°C inkubiert. Die Reaktion wurde durch Inkubation für 10 Minuten bei 75°C und 5mM EDTA gestoppt. 2.2.2.5. Auffüllung von DNA-Enden mit Hilfe von Klenow Die DNA wurde mit 40µM von jedem dNTP und 1U Klenow/µl DNA für 15 Minuten bei 25°C inkubiert. Durch Zugabe von EDTA mit einer Endkonzentration von 10mM und erhitzen auf 75°C für 10 Minuten, wurde die Reaktion gestoppt. 2.2.2.6. Ligation von DNA Für den Ligationsansatz wurde ein ungefähres Insert : Vektor-Verhältnis von 3 : 1 eingesetzt. Jeweils 1µl der drei Ligations-Lösungen ( siehe 2.1.2. unter Ligationsmix) und 1µl T4 DNA Ligase wurde dem Ansatz zugegeben und mit ddH2O auf ein Gesamtvolumen von 10µl gebracht. Der Ligationsansatz wurde über Nacht bei 16°C inkubiert. Nach Präzipitation des Ansatzes wurde die DNA in einem Volumen von 4µl aufgenommen und ca. 1µl davon zur Transformation in kompetente Zellen verwendet. 2.2.2.7. Präparation von elektrokompetenten Zellen Die Methode zur Präparation von elektokompetente Zellen basiert auf dem Protokoll von Dower et al. (1). Die DH10B Zellen wurden steril aus einem Glycerolstock entnommen und auf einer LB Platte ausgestrichen. Diese Platte wurde über Nacht bei 37°C inkubiert. Eine Kolonie wurde der Platte entnommen und erneut über Nacht bei 37°C in 20ml LB-Medium inkubiert. 10ml der Übernachtkultur wurden am folgenden Tag in 1l Terrific broth Medium (siehe 2.1.2.) auf einem Schüttler bei 37°C bis zu einer OD600 von 0,5 inkubiert. Die Zellen wurden anschließend für 30 Minuten auf Eis gestellt und schließlich bei 4000rpm in einem Sorvall GS3 Rotor für 15 Minuten bei 4°C zentrifugiert. Das Zellpellet wurde in 500ml kaltem Millipor-Wasser resuspendiert und erneut für 30 Minuten auf Eis gestellt. Nach einer weiteren Zentrifugation bei 8000rpm für 20 Minuten im 4°C kaltem Servall GS3 Rotor, wurde der Überstand abgeschüttet und das Pellet in ca. 250ml gekühltem Millipor-Wasser aufgenommen. Nach erneutem zentrifugieren bei 8000rpm wurde das Pellet in 10ml 10%Glycerol aufgenommen und in einer Heraeus-Zentrifuge bei 4000rpm für 7 Minuten und 4°C zentrifugiert. Der Überstand wurde vorsichtig abgegossen und das Zellpellet in 1ml 10%Glycerol aufgenommen. Die nun kompetenten Zellen wurden in 40µl Aliquots abgefüllt, in Flüssigstickstoff schock gefroren und bei -70°C gelagert. 2.2.2.8. Transformation Die kompetenten DH10B Zellen (siehe 2.2.2.7.) werden mit Hilfe der Elektotransformation transformiert. Hierfür werden 0,5-1µl der Plasmid DNA in den 40µl Aliquot kompetenter Zellen gegeben. Diese Lösung wurde in eine „Gene Pulser Cuvette“ (Biorad) pipettiert, in den Gene Pulser (Biorad) gespannt und 250kV Strom angelegt. Direkt nach der Transformation wurden die Zellen in 12 1ml LB-Medium aufgenommen und für eine Stunde bei 37°C auf dem Schüttler inkubiert. Nach der Inkubation wurden die Zellen abhängig von ihrer kompetenz bis zu 1 : 500 verdünnt und 100µl der Verdünnung auf eine LB-Agar Platte, die Ampicillin für die Selektion enthielt, ausgestrichen. Die Platte wurde über Nacht bei 37°C inkubiert. 2.2.2.9. Isolation von Plasmid-DNA Eine Kolonie transformierter Zellen wurde in 2ml LB-Medium mit Ampicillin bei 37°C über Nacht auf dem Schüttler inkubiert. 1,5ml der Kultur wurden am folgenden Tag bei 14000rpm für 5 Minuten in einer Tischzentrifuge (Eppendorf) zentrifugiert. Der Überstand wurde verworfen und das Pellet in 200µl Resuspensionspuffer P1 gelöst. Der Suspension wird 200µl Lysispuffer P2 zugegeben und 5 Minuten bei Raumtemperatur inkubiert. 200µl Neutralisationspuffer P3 wird zugegeben und die Suspension sofort gemischt. Anschließend wurde die Suspension für 15 Minuten bei 14000rpm und 4°C zentrifugiert, der Überstand wurde entnommen und zu 200µl Bindungssuspension gegeben. Das Ganze wurde in „Wizard mini colums“(Promega) pipettiert, die an eine Vakuumpumpe angeschlossen waren. Nachdem das Lysat durch den „mini colum“ gelaufen ist, wird der Filter mit 2ml Waschpuffer gewaschen und anschließend durch den Luftstrom der Vakuumpumpe getrocknet. Die Plasmid-DNA wurde durch Zugabe von 50µl 60°C warmen TEPuffer und vorsichtigem Zentrifugieren bei ca. 8000rpm von der Matrix eluiert. 2.2.3. Keimbahntransformation von Drosophila melanogaster P-Elemente sind eine von mehreren Transposon-Familien, die in Drosophila vorkommen. Sie erwiesen sich aufgrund ihrer Eigenschaften für genetische Experimente bei Drosophila als besonders nützlich. In der Natur liegen sie in vielen Individuen mit 30-50 Kopien unregelmäßig im Genom verstreut vor. Diese Drosophila-Individuen werden dem sogenannten P-Stamm zugeordnet und vom M-Stamm, der keine P-Elemente trägt, unterschieden. Wildtypische P-Elemente besitzen eine Länge von 2907 bp. An den Enden haben sie eine invertierte, exakte Sequenzwiederholung von 31 bp, die für Sprungereignisse essentiell ist. Diese „inverted repeats“ flankieren ein Gen, das für das Enzym Transposase kodiert und aus vier Exons besteht (Karess und Rubin, 1984); (Laski et al., 1986). Die Aktivität der Transposase ist für die Durchführung der Transposition notwendig. In Nachkommen einer Kreuzung von M-Stamm Weibchen und P-Stamm Männchen treten Transpositionen auf, wobei sich zeigte, daß die Transpositionsereignisse keimbahnspezifisch stattfinden. In somatischen Zellen wird das Transposase-Gen zwar expremiert, aber desssen 3. Intron nicht herausgespleißt, so daß es dort keine Transposase-Aktivität gibt. Die Eigenschaften der P-Elemente ermöglichen es, mit Hilfe von DNA-Injektionen transgene Fliegen zu erzeugen (Rubin und Spradling, 1982). Für die DNA-Injektion müssen Embryonen verwendet werden, die keine P-Elemente enthalten und somit dem M-Stamm angehören. Nach Injektion von transpositionsfähiger DNA (z.B. pUAST-Derivate) und in Gegenwart von Transposase, die hier durch P∆2-3 geliefert wurde, können stabile DNA-Integrationen in die Keimbahn der Empfänger entstehen und somit an einen Teil der Nachkommen vererbt werden. Um transformierte Fliegen in der F1-Generation zu erkennen, trägt die integrierte DNA einen Selektionsmarker (im Falle von pUAST das white-Gen). Es wird also durch die P-Element vermittelte Keimbahntransformation auch ein intaktes white-Allel in white--Embryonen eingeführt, so daß in transgenen Fliegen der G1-Generation eine Rettung des white-Phänotyps stattfindet. 2.2.3.1. Mikroinjektionsapparatur Die Mikroinjektionsapparatur bestand aus einem Nadelhalter, der in einen Mikromanipulator eingespannt war. Dieser erlaubt sehr feine Justierungen der Nadel in drei Dimensionen. Der Nadelhalter war mit einem Plastikschlauch verbunden, durch den geregelt Druckluft geleitet werden konnte. Durch ein in den Plastikschlauch integriertes T-Stück strömte die Luft ins Freie. Durch das Verschließen dieser Öffnung mit dem Finger wurde die Luft in Richtung Nadel umgeleitet und preßte dadurch die zu injizierende DNA-Lösung aus der Nadel hinaus. Am Mikroskop befand sich ein Objektträger, der in horizontaler Ebene in X- und Y-Richtung bewegt werden konnte. Ein Deckglas mit darauf festgeklbten Embryonen wurde auf einem Objektträger befestigt, der seinerseits in die Haltevorrichtung eingelegt wurde. Die Embryonen wurden durch eine Schicht Voltalev Öl 10S vor dem Austrocknen geschützt. Unter mikroskopischer Kontrolle wurde die Nadel in die gleiche Fokusebene wie die Embryonen gebracht. Diese wurden durch die Bewegung des Objektträgerhalters in die nun feststehende Nadel gedrückt und die zu 13 injizierende Lösung wurde durch das Schließen des Luftstromes mit dem Finger in den Embryo injiziert. 2.2.3.2. Ausziehen der Injektionsnadel Mikrokapillaren mit Filament (1mm Durchmesser) wurden in einen Nadelzieher eingespannt und fein ausgezogen. Die äußerste, verschlossene Spitze wurde kurz vor Gebrauch mit Hilfe einer Pinzette abgebrochen. 2.2.3.3. Klebstoff In einem 50ml Falcontube wurden eng gepackte Tesapack-Schnipsel in Heptan für mehrere Tage bei Raumtemperatur geschüttelt. Die Kleberlösung wurde anschließend von den Schnipseln befreit und kurz abzentrifugiert um die Schwebeteilchen aus dem Kleber zu entfernen. Der so entstandene Heptan-Kleber konnte im Kühlschrank aufbewahrt werden. Vor dem Bestreichen der Deckgläser (10 X 50 mm) wurde der Kleber kräftig geschüttelt und ca. 10µl an einer Seite des Deckglasrandes verteilt. 2.2.3.4. Dechorionierung von Embryonen Die Embryonen werden in Legekäfigen wie unter 2.2.1.1. beschrieben gesammelt. Zum anschließenden Dechorionieren der Embryonen wurde auf die Agarplatte 1 : 1 mit Wasser verdünntes Klorix gegeben. Nach etwa zwei Minuten haben sich die Embryonen aus ihren Eihüllen gelöst und wurden zur Oberfläche aufgetrieben. Die Embryonen wurden nun in einen kleinen Zylinder überführt, an dessen Unterseite ein feinmaschiges Kunstfasernetzchen befestigt war. Unter schwach fließendem Leitungswasser wurden die Embryonen zwei Minuten gespült und anschließend vorsichtig durch absaugen mit Hilfe eines Klinex-Tuches von der Unterseite des Netzes getrocknet. Im weiteren wurden sie wie unter 2.2.3.5. beschrieben wird behandelt 2.2.3.5. Allgemeine Vorarbeiten, Ausrichten der Embryonen und Injektion Dechorionierte white--Embryonen (siehe 2.2.3.4.), die etwa 30 Minuten alt waren und bei denen noch keine Polzellen zu sehen waren, wurden bei auf einen zuvor präparierten ApfelsaftagarStreifen übertragen. Unter einem Stereomikroskop (Okular 10x, Objektiv 3,2x) wurden die Embryonen mit Hilfe einer Nadel so ausgerichtet, daß ihre Längsachse in etwa parallel zur Querkante des Agarstreifens stand. In Abhängigkeit von verschiedenen Parmetern wie z.B. der Tageszeit, der Anzahl und des Alters der Weibchen konnten pro Gelege 100-150 Embryonen aufgereiht und für eine Injektionsserie verwendet werden. Durch vorsichtiges Andrücken der klebrigen Seite eines zuvor präparierten Deckglases (siehe 2.2.3.3.) auf die afgereihten Embryonen konnten dieselben vom Agarstreifen abgelöst werden. Anschließend wurden die Embryonen in einem Exsikkator bei Raumtemperatur für 8 bis 10 Minuten getrocknet. Unmittelbar danach wurden die Embryonen mit 10S Voltalevöl überschichtet, um sie für den Injektionsvorgang transparenter zu machen und sie vor dem Austrocknen zu schützen. Während des Trocknens wurde die zuvor vorsichtig abgebrochene Nadel (siehe 2.2.3.2.) mit der zu injizierende DNA-Lösung beladen. Hierfür wurde der Luftzufuhr zur Injektionsnadel abgedreht und eine 50ml Spritze an das T-Stück im Schlauch angeschlossen (siehe 2.2.3.1.). Die zu injizierende DNA-Lösung wurde in einer Tropfengröße von ca. 4µl auf ein Silikonisiertes Deckgläschen Pipetiert. Die Nadelspitze wurde in die DNA-Lösung eingetaucht und durch Erzeugung eines Unterdrucks, mit Hilfe der Spritze, wurde die Lösung in die Nadel gesogen. Die Endkonzentration der DNA-Lösung betrug 0,5µg/µl für die pUAST-DNA und 0,1µg/µl für das Helferplasmid P ∆2-3 (siehe 2.2.3.). Es wurden schätzungsweise 0,5-1,0 nl DNA-Lösung pro Embryo in die Region des Polplasmas injiziert. 2.2.3.6. Identifizierung transgener Fliegen Nach der Injektion wurde das Deckglas mit den Embryonen in eine kleine Plastikschale gelegt und vollständig mit 3S Voltalevöl überschichtet, um ein Austrocknen der Embryonen zu verhindern. Daraufhin wurden die Embryonen bis zum Schlüpfen bei Raumtemperatur in einer feuchten Kammer aufbewahrt. Die geschlüpften Larven wurden in Futtertöpfen, die zusätzlich mit frischer Hefe versehen waren, überführt. Jedes geschlüpfte Männchen wurde mit 3 white--Jungfrauen und jedes geschlüpfte Weibchen (möglichst als Jungfrau) mit 3 white--Männchen gekreutzt. Die 14 Nachkommenschaft aus diesen Einzelkreuzungen wurden auf das Auftreten von white+-Fliegen analysiert. Traten solche transgenen Fliegen auf, wurden diese wiederum mit white--Fliegen des anderen Geschlechtes weiter gekreutzt, um Die transgenen Fliegen für weitere Experimente zu expandieren. 2.2.4. In vitro Transkription für RNA Injektionen Die Plasmide wurden für zwei Stunden mit EcoR I linearisiert. 2µg der linearisierten Plasmide wurden in einer in vitro Transkription eingesetzt. Ein 20µl Ansatz bestand aus: 4µl 5x Transkriptionspuffer (200mM Tris/HCl pH 7,5, 30mM Mg2Cl2, 10mM Spermidine), 2µl DTT (0,1M), 1µl RNasin (20U/µl), 4µl 5x rNTPs (ATP, UTP, CTP je 2,5mM, 2,5mM m 7G(5’)ppp(5’)G, 0,5mM GTP) und 5U SP6 Polymerase. Nach einer Stunde Inkubation bei 37°C wurde 2µl GTP (5mM) zugegeben und für weitere 30 Minuten bei 37°C inkubiert. Die RNA wurde phenolisiert, mit Ethanol gefällt und in soviel Millipor-Wasser aufgenommen, daß man eine RNA-Konzentration von 1,5µg/µl erhielt. 2.2.4.1. RNA Injektion in Embryonen von Drosophila melanogaster Die Injektion von RNA in Embryonen entspricht der DNA Injektionen und wurde in 2.2.3. beschrieben. Für die RNA Injektionen wurden keine Embryonen von white--Fliegen, sondern die von white+-Fliegen verwendet. 2.2.4.2. Standardfixierung und Entvitellinisierung Die dechorionierten und injizierten Embryoenen wurden mit Heptan vom Deckgläschen gelöst und von dem 10S Voltalevöl befreit. Anschließend wurden sie in eine 1 : 1 Mischung aus 700µl Heptan und 700µl 4% Formaldehydlösung in PBT (frisch angesetzt) aufgenommen. Die Embryonen wurden in dieser Fixierlösung 30 Minuten bei Raumtemperatur unter ständiger leichter Rotation inkubiert. Anschließend wurde die untere Phase mit einer Pasteurpipette entfernt und die in der Interphase schwimmenden Embryonen auf ein Gitterchen pipettiert. Die Fixierflüssigkeit wurde durch ein saugfähiges Papiertuch unter dem Gitterchen abgesaugt. Die Embryonen wurden vorsichtig auf ein Stück Doppelklebeband überführt und in eine kleine Schale geklebt, so daß sich die Embryonen oben auf dem Klebestreifen befinden. In die Schale wurde PBS gegeben bis die Embryonen bedeckt waren. Unter dem Stereomikroskop wurden die Embryonen mit einer feinen Nadel von der Vitellinmembran befreit. Die entvitellinisierten Embryonen wurden in Methanol aufgenommen und konnten so bei -20°C gelagert werden. 15 3. Ergebnisse 3.1. Analyse der Stabilität des Cyclin A In den ersten 14 Kernzyklen von Drosophila zeigen Cyclin A und Cyclin B unterschiedliche Stabilität. Ab Zyklus 14 ist sowohl Cyclin A wie auch Cyclin B in der Interphase stabil. Während der Mitose und der gesamten G1-Phase sind Cyclin A und Cyclin B instabil. Diese Instabilität von Cyclin A und B wird verursacht durch einen destruction box abhängigen Abbau. Proteine, die eine destruction box besitzen, werden vom APC als Substrat zur Ubiquitinierung erkannt (Murray et al., 1989;Glotzer et al., 1991; Kobayashi et al., 1992; Lorca et al., 1992; Amon et al., 1994). Diese Ubiquitinierung vermittelt die rasche proteolytische Degradation der auf diese Weise markierten Proteine durch das sogenannte 26S Proteasom (Gordon et al., 1993; Richter-Ruoff and Wolf, 1993). Diese Proteolyse der Cycline führt zur Inaktivierung der Cdk1 Kinaseaktivität und ermöglicht den Austritt aus der Mitose. Beim Eintritt in die S-Phase wird der APC inaktiviert und ermöglicht die Akkumulierung der mitotischen Cycline. In diesen frühen Zyklen ist das Cyclin A sowohl in der Interphase wie auch während der Mitose instabil. Welcher Mechanismus allerdings für die Instabilität von Cyclin A verantwortlich ist und wie die Regulation dieser frühen Kernzyklen erfolgt, ist noch unklar. Ob während der Kernzyklen 2-7 der destruction box abhängige Abbau von Cyclin A eine Rolle spielt, ist noch nicht bekannt. Ob die Instabilität von Cyclin A in den ersten Zellzyklen destruction box abhängig erfolgt, wurde mit Hilfe verschiedene Konstrukte (Abb. 2) von Cyclin A und B untersucht. Sequenzvergleiche der destruction box zeigen ausgeprägte Homologie und erlauben die Ableitung einer Konsensussequenz, die nachfolgend den korrespondierenden Sequenzen von Cyclin A und Cyclin B gegenübergestellt ist (King et al., 1996). Auffallend ist die abweichende Lage der destruction box in der Primärsequenz beider Proteine. 1 2 3 4 5 6 7 8 9 Konsensus R X X L X X X X N für B-Typ Cycline Cyclin B R A A L G D L Q N Position: 37 Cyclin A R S I S Position: 160 L G V I Q Konsensus R X X L X X X X X Für A-Typ Cycline Abb.1.: Die Aminosäuresequenz von Cyclin A und Cyclin B in Drosophila melanogaster. Die Konsensussequenz von Cyclin A unterscheidet sich in einer konservierten Aminosäure an Position 9 von der Konsensussequenz von Cyclin B. Das Arginin an Position 1 ist die am stärksten konservierte Aminosäure der destruction box. 16 Ein destruction box abhängiger Abbau kann durch komplette Deletion der destruction box am N-Terminus verhindert werden. Bei Cyclin A fehlen dem Protein dadurch die N-terminalen 169 Aminosäuren. Die Funktion dieser 169 Aminosäuren für die Stabilität von Cyclin A ist, abgesehen von der destruction box, noch nicht bekannt. In späteren Zellzyklen führt die Expression von Cyclin A mit deletiertem N-Terminus (Cyclin A∆169), einschließlich der destruction box, zu einem Block in der Mitose ( Sigrist et al., 1995). Weiterhin wurde sowohl in vivo als auch in vitro experimentell gezeigt, daß dem Cyclin A∆169 die Funktion als Cyclin-Partner von Cdk1 erhalten bleibt (Foley, Diplomarbeit 1998; Sprenger, 1997). Ein weiteres Konstrukt von Cyclin A wurde hergestellt, bei dem die Gesamtlänge des Proteins erhalten bleibt, jedoch wurde die Funktion der destruction box durch eine Punktmutation beeinflußt. Die am stärksten konservierte Aminosäure der destruction box (Arg→Gly) wurde ausgetauscht. In anderen Organsimen wurde die Stabilität von Cyclin B, mit einer Mutation des Arginins in der destruction box, schon mit Erfolg nachgewiesen (Glotzer, 1991; Kobayashi et al., 1992; Lorca et al., 1992; Steward et al., 1994). Für das Drosophila Cyclin A und B konnte dies allerdings noch nicht gezeigt werden. Die Cyclin A Konstrukte wurden darüberhinaus mit einer Haemagglutinin-Epitop Sequenz aus dem Influenza Virus (HA-tag) versehen, um die Cycline über anti-HA-Antikörper vom endogenen Cyclin unterscheiden zu können. Zur Analyse der funktionellen Bereiche von Cyclin A wurden die N-terminalen 169 Aminosäuren separat mit HA-tag in vitro transkribiert/translatiert und auf ihre Stabilität hin untersucht. Alle diese Konstrukte befinden sich in einem SP64 abgeleiteten Vektor (Siegel and Walter, 1988) mit einer nicht translatierten 5’ Region vom β-Globin Gen aus Xenopus. Der SP64 Vektor ermöglichte eine in vitro Transkription der Konstrukte. Die erhaltene RNA wurde in späteren Experimenten (3.2.) in Embryonen injiziert oder für Stabilitätsuntersuchungen in vitro translatiert. Die β-Globin Sequenzen ermöglichen hierbei eine bessere in vitro Translation. 17 Cyclin A Cyclin A∆169 Cyclin AR160G Cyclin B Cyclin B∆46 HA- Cyclin A HA- Cyclin A∆169 HA-Cyclin AR160G HA-169N-Terminale AS von Cyclin A Abb. 2.: Cyclin A und Cyclin B Konstrukte. Alle Konstrukte besitzen eine sogenannte Cyclin-Box ( ). Diese Cyclin-Box stellt den Bereich des Proteins dar, mit dem die Kinase assoziiert. Die destruction box ( ) ist eine Domäne die für die proteolytische Degradation der Cycline verantwortlich ist. Durch Deletion wurden Konstrukte erzeugt, die keine destruction box mehr besitzen. Diese Deletion sollte einen destruction box abhängigen Abbau verhindern. Um einen Einfluß auf die Stabilität und Funktion von Cyclin A durch das Fehlen der ersten 169 Aminosäuren auszuschließen, wurde ein Cyclin A mit nur einem Aminosäureaustausch an Position 160 der destruction box hergestellt. Hierfür wurde die Aminosäure R (Arg), die 100%ig konserviert ist, an Position 160 durch Gly ausgetauscht. Dem Cyclin B fehlen durch Deletion 46 Aminosäuren am N-Terminus. Alle Cyclin A Konstrukte wurde zusätzlich mit einem HA-tag ( ) versehen, um die Cyclin A Konstrukte vom endogenen Cyclin A unterscheiden zu können. 18 3.1.1. Das mit HA-Cyclin A und HA-Cyclin A ∆169 assoziierte Cdk1 (cdc2) zeigt im Kinase-Assay Aktivität Durch die Komplexbildung zwischen dem Cyclin A und Cdk1 wird auch die Stabilität von Cyclin A beeinflußt. Es muß also sichergestellt werden, daß sowohl das HA-Cyclin A wie auch das HA-Cyclin A∆169 trotz HA-tag und/oder Verlust von 169 Aminosäuren am N-Terminus, noch in der Lage sind mit Cdk1 einen Komplex zu bilden. Dies kann durch eine Immunpräzipitation untersucht werden. Die HA-Cyclin A Konstrukte wie auch Cdk1 wurden in vitro transkribiert und mit 35S-Met im Retikulozyten Lysat translatiert. Die in vitro translatierten Proteine wurden im Immunpräzipitationspuffer inkubiert und die HA-Cyclin A Konstrukte mit Hilfe eines an Protein G-Agarose beads gekoppelten HA-Antikörpers aus dem Extrakt präzipitiert. Im anschließenden Kinase-Assay wurden die Aktivität des komplexierten Cdk1 analysiert. Die Cdk1-Kinase muß durch Phosphorylierung am T161 (siehe 1.) aktiviert werden. Dies geschieht durch Zugabe von CIV, einer Kinase aus Saccharomyces cerevisiae. Diese CIV Kinase phosphoryliert Cdk1 am T161. Dadurch wir Cdk 1 aktiviert und phosphoryliert die als Substrat angebotenen H1 Histone unter Zugabe von. γ32-ATP. Über ein Polyacrylamidgel wurden anschließend die Proteine aufgetrennt. Dieser Kinase Assay wurde von Edan Floley im Rahmen seiner Diplomarbeit etabliert und mit den hier verwendeten Cyclin Konstrukten durchgeführt. Wurde dem Versuchsansatz kein Cyclin A zugefügt, erhält man nur eine geringe Phosphorylierung der H1 Histone (Abb. 3 Spur 1). Diese Phosphorylierung kann als Background interpretiert werden. Inkubiert man dagegen die in Retikulozyten Lysat translatierten HA-Cyclin A Konstrukte nur mit GST-CIV erhält man ein starkes Phosphorylierungssignal (Abb. 3, Spur 2 und 3). Diese Phosphorylierung der Histone kommt durch das Vorhandensein von Cdk1 im Retikulozyten Lysat zustande. Dieses Cdk1 assoziiert mit den HA-Cyclin A Konstrukte und wird durch das GST-CIV aktiviert. Eine Inkubation von HA-Cyclin A Konstrukte mit im Retikulozytenlysat translatiertem Drosophila Cdk1 zeigt unter diesen Bedingungen ein vergleichbares Phosphorylierungssignal (Abb. 3 Spur 4 und 5). Dieser Kinase-Assay hat also gezeigt, daß sowohl die mit HA-Cyclin A wie auch mit HA-Cyclin A∆169 assoziierte Cdk1 Kinase aktiv ist. Das heißt, das HA-tag und auch die fehlenden N-Terminalen Aminosäuren, haben keinen Einfluß auf die Funktion von Cyclin A. 19 Abb. 3: Kinase-Assay. Reticulozyten translatiertes 35S markiertes HA-Cyclin A und HA-Cyclin A∆169 (HACyclin Ad169) wurde mit Cdk1 und GST-CIV in Anwesenheit von ATP inkubiert. Die HA-getagten Cycline wurden mit Hilfe von αHA Antikörpern präzipitiert und mit H1 Histonen als Substrat und γ32-ATP inkubiert. Die Auftrennung der Proteine erfolgte durch SDS-Polyacrylamidgel-Elektrophorese. Die Kontrollen in Spur 1 zeigt ohne Zugabe von Cyclin A eine schwache Bande, die als „background“ betrachtet werden kann. Das Reticulozyten Lysat, welches zur in vitro Translation verwendet worden ist, beinhaltet endogenes Cdk1. Dieses Reticulozyten Cdk1 assoziiert mit den HA-Cyclin A Konstrukten, es wird durch GST-CIV aktiviert und phosphoryliert die als Substrat angebotenen H1 Histone (Spur 2 und 3). Inkubiert man HA-Cyclin A und HA-Cyclin A∆169 mit dem Substrat, erhält man ein starkes Phosphorylierungssignal der Histone (Spur 4 und 5). 20 3.1.2. In vitro Analyse zur Stabilität von Cyclin A Während der ersten Zyklen erfolgt eine ständige Translation von Cyclin A aus der im Embryo vorhandenen maternalen mRNA. Dieses Cyclin A ist während dieser frühen Kernteilungen instabil. Der Abbau des Cyclin A wird durch die Synthese vollständig ausgeglichen, so daß die Cyclin A Menge im Embryo immer gleich bleibt. Das Cyclin B ist in den frühen Kernzyklen ebenfalls in gleichbleibender Menge vorhanden. Im Gegensatz zum Cyclin A ist das Cyclin B in den ersten 8 Kernzyklen stabil. Zur Untersuchung der Mechanismen, die die Stabilität von Cyclin A und B beeinflussen, wurde ein in vitro System etabliert. Dieses in vitro System ermöglicht es die Stabilität der in vitro translatierten Cyclin Konstrukte zu untersuchen. Dazu wurde aus 0-1 Stunden alten Drosophila-Embryonen ein Extrakt hergestellt (2.2.1.2.). Dieser Extrakt beinhaltet die löslichen Bestandteile der Embryonen vom Kernzyklus 1 bis 7. Die verschiedenen Cyclin A und B Konstrukte wurden in vitro transkribiert (2.2.1.3.) und anschließend im Retikulozyten Lysat translatiert (2.2.1.4.). Die Analyse der Stabilität dieser Konstrukte erfolgte anschließend im Embryonenextrakt. Auf ein Volumen von 50µl Extrakt kamen 2mM ATP, 10mM MgCl 2 und 20µl der in vitro translatierten Konstrukte. Die Inkubation erfolgte bei 25°C. Nach den angegebenen Zeitintervallen wurden 7µl (1/10 des Volumens) Extrakt entnommen und mit Lämmli Puffer (2.1.11.) inaktiviert (2.2.1.5.). Die Proteine wurden zunächst im SDS-Polyacrylamidgel (SDS-PAGE) (2.1.11.) aufgetrennt, auf Nitrozellulose geblottet und mit Hilfe von Antikörpern (2.2.1.6.) bzw. durch Autoradiographie nachgewiesen. Zunächst wurde die Stabilität der Konstrukte mit und ohne destruction box (HA-Cyclin A, HA-Cyclin A∆169) im Embryonenextrakt getestet. Im Vergleich von HA-Cyclin A und HA-Cyclin A∆169 zeigt sich nach 45 Minuten ein vollständiger Abbau des HA-Cyclin A, während das verkürzte Cyclin A (HA-Cyclin A∆169) stabil bleibt. Das HA-Cyclin A zeigt nach 15 minütiger Inkubation einen leichten Abbau und es erscheint eine zweite höhermolekulare Bande. Es könnte sowohl eine Phosphorylierung des Cyclin A wie auch der Transfer eines Ubiquitinrestes als Ursache für dieser „Verschiebung“ in Frage kommen. Auch das HACyclin A∆169 zeigt diese zusätzliche höhermolekuare Bande. Daher scheint diese Art der Modifikation unabhängig von den N-Terminalen 169 Aminosäuren zu erfolgen (Abb. 4 A). Das HA-Cyclin AR160G wird wie das HA-Cyclin A schon nach 45 Minuten vollständig abgebaut (Abb. 4 B). Das HA-Cyclin AR160G zeigt also in diesem Experiment nicht die gleiche Stabilität, wie sie bei dem HACyclin A ∆169 zu erkennen ist. Dies läßt vermuten, daß der Abbau des Cyclin A im Embryonenextrakt nicht destruktion box abhängig erfolgt, der Aminosäureaustausch die destruktion box nicht vollständig inaktiviert oder, daß eine zweite destruction box vorhanden ist. 21 Um die Stabilität dieser N-Terminalen 169 Aminosäuren im Extrakt zu testen, wurde ein Haemagglutinin markiertes Konstrukt hergestellt, welches nur diese 169 Aminosäuren beinhaltet, die dem HA-Cyclin A∆169 fehlen. Dieses Konstrukt wird kontinuierlich über die getesteten 70 Minuten hinweg abgebaut (Abb. 4 C). Die Anwesenheit dieser N-terminalen Sequenz scheint daher für die Instabilität von Cyclin A verantwortlich zu sein. Abb. 4: Stabilität der Cyclin A Konstrukte in 0-1 Std. altem Embryonenextrakt. Die Stabilität der verschiedenen HA markierten Cyclin A Konstrukte wurde nach Auftrennung über ein 10%iges SDS-PAGE, mittels Western-Blots nachgewiesen. Die in vitro translatierten Konstrukte wurden in 0-1 Stunde altem Embryonenextrakt bei 25 C° inkubiert und in den angegebenen Zeitintervallen wurden Proben entnommen. A: Western-Blot mit HA-Cyclin A und dem verkürzten HA-Cyclin A∆169. Das HA-Cyclin A wird nach einer Inkubationszeit von 45 Minuten vollständig abgebaut. Das HA-Cyclin A∆169 verhält sich, über den gemessenen Zeitraum von 70 Minuten, stabil. B: Western-Blot mit HA-Cyclin AR160G und HA-Cyclin A∆169 . Das HA-Cyclin AR160G ist nach 45 Minuten vollständig abgebaut. C: Western-Blot mit den N-Terminalen 169 Aminosäuren von Cyclin A mit einem Haemagglutinin Fusionsprotein. Die N-Terminalen 169 Aminosäuren von Cyclin A werden während der gemessenen Zeit von 70 Minuten kontinuierlich abgebaut. Da im Western Blot nur das HA-tag nachgewiesen wurde und somit nicht der Abbau der Cyclin Konstrukte direkt beobachtet werden kann, wurden alle Konstrukte in vitro mit 35 S-Met markiert. Durch dieses Markierungsverfahren sind sowohl die Modifikationen wie auch die Abbauprodukte besser zu erkennen. 22 ∆169 Das HA-Cyclin A wie auch das HA-Cyclin A zeigen nach einer Inkubationszeit von 15 Minuten eine höhermolekulare Bande, die schon im Western-Blot (Abb. 4 A) beobachtet wurde. Nach 45 Minuten ist HACyclin A vollständig abgebaut und es treten Banden höherer Mobilität (durch * gekennzeichnet) auf (Abb. 5 A). Das beobachtete Abbaumuster wurde in verschiedenen Versuchsansätzen beobachtet. Der Abbau von Cyclin A erfolgt daher nicht unspezifisch, sondern wird durch eine definierten Proteaseaktivität hervorgerufen. Auch das HA-Cyclin AR160G wird in gleicher Weise abgebaut. Bei dem stabilen Cyclin A (HA-Cyclin A∆169) ist unter diesen Bedingungen auch eine höhermolekulare Bande sichtbar, das Protein bleicht jedoch stabil. Bei den niedermolekularen Banden, die schon nach 0 Minuten Inkubation zu erkennen sind, handelt es sich um verkürztes Cyclin A. Dieses verkürzte Cyclin A findet man auch in Retikulozyten Lysat translatierten Proben die keinen Embryonenextrakt beinhalten. Abb. 5: Autoradiogramme zum Nachweis der Stabilität der HA-Cyclin A Konstrukte in 0-1 Std. altem Embryonenextrakt. Retikulozyten translatierte 35S markierte Cyclin A Konstrukte wurden für 0, 15, 45 und 70 Minuten in 0-1 Stunde altem Embryonenextrakt inkubiert. Die Proteine der entnommenen Proben wurden mittels 10%igem SDS-PAGE aufgetrennt. A: Autoradiogram zur Darstellung der Stabilität von HA-Cyclin A und dem verkürzten HA-Cyclin A∆169. Der Abbau des HA-Cyclin A bringt reproduzierbar ein Fragment (*) hervor. B: Autoradiogram zur Darstellung der Stabilität von HA-Cyclin AR160G und HA-Cyclin A∆169. Auch der Abbau von HA-Cyclin AR160G führt reproduzierbar zu einem Fragment (*). 23 35 Im nächsten Versuch wurde mit S-Met markiertes Cyclin A und Cyclin A ∆169 eingesetzt, um auszuschließen das das HA-tag den Abbau von Cyclin A beeinflußt. Wie in Abb. 6 A zu sehen ist, zeigt der Abbau von Cyclin A das gleiche Bandenmuster wie das HA-Cyclin A in Abbildung 5 A. Reproduzierbarkeit und Intensität der einzelnen Banden im Vergleich zeigen, daß das HA keinen nachweisbaren Einfluß auf die Stabilität der Cycline hat. Das Cyclin A∆169 bleibt während des getesteten Zeitraums stabil (Abb.6 B). In den bisherigen Versuchen konnte beobachtet werden, daß eine Modifikation von Cyclin A mit anschließendem Abbau erfolgte. Beim destruction box abhängigen Abbau erfolgt eine Konjugation von Proteinen mit Ubiquitin. Der Polyubiquitinierung folgt die Proteolyse des Proteins. Auch konnte keine höhermolekulare Bande beim HA-Cyclin AR160G beobachtet werden (Abb. 7 B). Der Abbau von HA-Cyclin AR160G erfolgte jedoch unabhängig von der ausbleibenden Modifikation. Um zu untersuchen ob die Modifikation von Cyclin A und die Abbaurate durch Zugabe von Ubiquitin beeinflußt werden kann, wurden die Versuche in Gegenwart von 0,5mg/ml Ubiquitin wiederholt. Wie in Abb.7 zu sehen ist, hat die Zugabe von Ubiquitin keinen Einfluß auf die Abbaurate. Es ist jedoch bemerkenswert, daß die Intensität der höhermolekularen Bande zunimmt. Auch diese Beobachtung konnte in verschiedenen Ansätzen reproduziert werden. Abb. 6: Stabilität von Cyclin A und Cyclin A∆169 in 0-1 Stunde altem Embryonenextrakt. Hierfür wurden Retikulozyten translatierte 35S markierte Cyclin A Konstrukte ohne HA-tag verwendet. Die zugeführten Proteine wurden bei 25°C im Embryonenextrakt inkubiert, nach den angegebenen Zeitintervallen entnommen und über ein 10%iges SDS-PAGE getrennt. A: Das Cyclin A zeigt nach 15 Minuten die bereits bekannte höhermolekulare Bande. Nach 45 Minuten ist das Cyclin A vollständig abgebaut. B: Das Cyclin A∆169 wird im gemessenen Zeitraum nicht abgebaut. Nach 15 Minuten erscheint eine höhermolekulare Bande, die auf eine bereits beobachtete Modifikation schließen läßt. 24 Abb. 7: Die Zugabe von Ubiquitin führt zu keiner Veränderung des Cyclin A Abbaus. Dem Embryonenextrakt wurde 0,5mg/ml Ubiquitin zugefügt, um auszuschließen das Ubiquitin im Embryonenextrakt der limitierende Faktor für eine Polyubiquitierung ist. Analysiert wurde die Stabilität der zugesetzten Cycline mittels Western-Blot. Zur Kontrolle (Ko.) wurde in vitro translatiertes HA-Cyclin A, HA-Cyclin AR160G und HA-Cyclin A∆169 aufgetragen. A: HA-Cyclin A und HA-Cyclin A∆169 , mit und ohne Zugabe von 0,5mg/ml Ubiquitin. Die Anwesenheit von Ubiquitin läßt keinen Einfluß auf die Abbaurate von Cyclin A erkennen. B: HA-Cyclin AR160G und HA-Cyclin A∆169 , mit und ohne Zugabe von 0,5mg/ml Ubiquitin. Das HA-Cyclin AR160G wird nach Ubiquitinzugabe mit gleicher Geschwindigkeit und gleicher Intensität abgebaut. Dem HA-Cyclin AR160G fehlt die höhermolekulare Bande, die beim HA-Cyclin A zu erkennen ist. 25 3.1.3. In vitro Analyse zur Stabilität von Cyclin B Cyclin B enthält wie Cyclin A eine destruction box. Für Cyclin B ist in mehreren Organismen, einschließlich Drosophila, gezeigt worden, daß die Deletion der destruction box eine Polyubiquitinierung und einen daraus resultierenden Abbau durch das Proteasom, verhindert (Su, 1998; Sigrist, 1995). Um zu testen wie sich dieses Konstrukt in dem in vitro Assay verhält, wurden zusätzlich zum Cyclin A, Cyclin B und ein Cyclin B ∆46 in die Stabilitätsexperimente mit einbezogen. Dem Cyclin B ∆46 fehlen die 46 Nterminalen Aminosäuren einschließlich der destruction box. Cyclin B wird in 0-1 Stunden altem Embryonenextrakt nach 45 Minuten Inkubation abgebaut (Abb. 8 A). Im Vergleich zum Cyclin A (Abb. 6 A) ist hier keine Modifikation von Cyclin B zu erkennen. Aber auch hier findet nach 15 Minuten eine spezifische Fragmentierung von Cyclin B statt, welche in dem auftretenden Bandenmuster zu erkennen ist. Das Cyclin B∆46 wird in diesem in vitro Experiment trotz fehlender destruction box abgebaut (Abb. 8 B). Auch hier ist keine Modifikation von Cyclin B∆46 zu erkennen. Diese Ergebnisse lassen die Vermutung zu, daß dieser hier beobachtete Abbau nicht destruction box abhängig erfolgt. Der Abbau kann aber durchaus als Resultat spezifischer Proteaseaktivität betrachtet werden. Abb. 8: Stabilität von Cyclin B und Cyclin B∆46 in 0-1 Stunde altem Embryonenextrakt. Hierfür wurden Retikulozyten translatierte 35 S markierte Cyclin A und Cyclin B Konstrukte ohne HA Fusionsanteil verwendet. Die zugeführten Proteine wurden bei 25 C° im Embryonenextrakt inkubiert, nach den angegebenen Zeitintervallen entnommen und über ein 10%iges SDS-PAGE getrennt. A: Das Cyclin B ist nach 45 Minuten teilweise und nach 70 Minuten vollständig abgebaut. Die mit einem * versehene Bande nimmt in gleichem Umfang zu, wie die Cyclin B Bande abnimmt. B: Das Cyclin B∆46 wird mit gleicher Rate abgebaut wie das Cyclin B. Bei den mit einem # versehenen Banden handelt es sich um stabiles Cyclin B. Cyclin B besitzt interne Trunkationsstellen. 26 3.1.4. Einfluß von Proteasomeninhibitoren auf den Cyclin-Abbau Die bisherigen Daten haben gezeigt, daß der Abbau der Cycline in einem in vitro System nicht destruction box abhängig erfolgt. Ein destruction box abhängiger Abbau konnte z.B. für Cyclin B in mitotischem Xenopus-Extrakt beobachtet werden (Vorlaufer and Peters; 1998). Durch Zusatz von ProteasomenInhibitoren wird dieser Abbau verhindert, und es kann ein charakteristisches Bandenmuster beobachtet werden. Dieses Bandenmuster kann auf die Ubiquitinaddition zurückgeführt werden. Welchen Einfluß der Einsatz eines Proteasomen-Inhibitors auf den Abbau der Cyclin Konstrukte in einem in vitro System hat wurde in den nachfolgenden Experimenten untersucht. Um die geeignete Konzentration zu ermitteln, wurden verschiedene Konzentrationen zweier in der Literatur beschriebenen Inhibitoren, Cbz-LLL (Carbobenzoxy-L-leucyl-L-leucyl-L-leucinal) und N-Ac-Leu-Leu-norleucinal (N-Acetyl-Leucyl-Leucylnorleucinal), getestet (Abb. 9). In Abb. 9B kann erkannt werden, daß eine partielle Inhibition des HA-Cyclin A Abbaus durch N-Ac-Leu-Leu-norleucinal bei einer Konzentration von 0,2mg/ml erfolgt. Eine 16fach geringere Menge an N-Ac-Leu-Leu-norleucinal genügt, um den Abbau von Cyclin B in einem XenopusExtrakt zu blockieren. Um eine fast vollständige Inhibition im Drosophila-Embryonenextrakt zu erhalten, müssen 2,4mg/ml N-Ac-Leu-Leu-norleucinal eingesetzt werden. Durch Cbz-LLL konnte nur eine geringe Inhibition erreicht werden. Auch konnte in beiden Fällen kein durch Ubiquitinaddition erzeugtes Bandenmuster erkannt werden. Abb. 9: Titration der Proteasomen-Inhibitoren Cbz-LLL und N-Ac-Leu-Leu-norleucinal. Es wurde pro eingesetzter Inhibitor-Konzentration nur ein 0 Minuten und ein 45 Minutenwert entnommen. Der 0 Minutenwert ist der Kontrollwert für die eingesetzte Cyclinmenge. Nach 45 Minuten ist das HA-Cyclin A ohne Inhibition des Abbaus bereits abgebaut. A: 0,8mg/ml N-Ac-Leu-Leu-norleucinal verhindert den Abbau von HA-Cyclin A. Bei dieser Konzentration wurde im Xenopusextrakt eine Inhibition des Cyclin B Abbaus erreicht. B: Keine der hier eingesetzten Konzentration an Cbz-LLL inhibiert den Abbau von Cyclin A vollständig. 27 Eine Inhibition des Cyclin A Abbaus wurde durch den Einsatz von 2,4 mg/ml N-Ac-Leu-Leu-norleucinal erreicht. In den nachfolgenden Experimenten wurden der Abbau von Cyclin A und B ohne HA-tag untersucht. Hierfür wurden die Cyclin A und B Konstrukte mit S35 markiert. Dies ermöglicht eine genauere Analyse des Abbaumusters und schließt einen möglichen Einfluß des HA-tag auf die Stabilität der Konstrukte aus. Der Abbau von Cyclin A wird durch 2,4mg/ml N-Ac-Leu-Leu-norleucinal nicht vollständig verhindert (Abb. 10 A). Der N-Ac-Leu-Leu-norleucinal behandelte Embryonenextrakt zeigt nach 15 minütiger Inkubation die bereits bekannte höhermolekulare Bande sowie ein vermutlich niedermolekulares Degradationsprodukt des Cyclin A. Bei weiterer Inkubation erhält man kein Polyubiquitinierungsmuster von Cyclin A und nur einen geringen Abbau als Resultat spezifischer aber limitierter Fragmentierung. Der Ansatz ohne N-Ac-Leu-Leu-norleucinal zeigt die bereits bekannten Fragmentation von Cyclin A in spezifische Fragmente. Das stabile Cyclin A∆169 zeigt weder eine Polyubiquitinierung noch einen Abbau (Abb. 10 B). Durch die Zugabe von N-Ac-Leu-Leu-norleucinal bleibt aber in diesem Ansatz auch die Modifikation des Cyclin A∆169 aus. Der Abbau der N-terminalen 169 Aminosäuren, mit intakter destruction box, wird durch den Einfluß von N-Ac-Leu-Leu-norleucinal stabilisiert (Abb.10 C). Hier ist keine Modifikation/Polyubiquitinierung nachweisbar. Auch der Abbau von Cyclin B und Cyclin B∆46, für welchen ein destruction box abhängiger Abbau in späteren Zellzyklen demonstriert wurde, wird in diesem Versuchsansatz durch N-Ac-Leu-Leu-norleucinal verlangsamt (Abb.10 D und E). Da eine Kontrolle für eine vollständige Inhibition der Proteasomen nicht verfügbar ist, bleibt derzeit noch offen, ob weiterhin ein Abbau durch Ubiquitin abhängige Proteolyse in geringem Maß erfolgt. Dieser Vorgang würde es in diesem Versuchsansatz unmöglich machen, eine schwache Modifikation durch Ubiquitin nachzuweisen. Die Daten können dahingehend interpretiert werden, daß eine Proteolyse der im Embryonenextrakt eingesetzten Cyclin Konstrukte durch N-Ac-Leu-Leu-norleucinal partiell verhindert werden kann. In Drosophila konnte in späteren Zellzyklen bereits gezeigt werden, daß Cyclin B∆46 nicht destruction box abhängig abgebaut werden kann (Su et al., 1998). Da dieses stabile Cyclin B∆46 im in vitro System abgebaut wurde, scheint dieser Abbau unabhängig von der destruction box zu erfolgen. Das Auftreten reproduzierbarer Fragmente beim Cyclin Abbau und die Inhibition des Cyclin-Abbaus durch N-Ac-Leu-Leu-norleucinal, spricht allerdings für einen spezifischen Abbau durch Proteasen. Die Inhibitorkonzentrationen, die hier eingesetzt wurden, sind jedoch viel höher als vergelichbare in der Literatur beschriebene Werte. Proteaseinhibitoren haben ein breites Wirkspektrum, und das hier wirksame N-Ac-Leu-Leu-norleucinal ist auch in der Lage Caspasen zu inhibieren (Yoshimura, 1983; Squir, et al., 1994). 28 29 Abb. 10: Wirkung von 2,4 mg/ml N-Ac-Leu-Leu-norleucinal auf den Abbau verschiedener Cyclin A und Cyclin B Konstrukte. Es wurden Retikulozyten translatierte 35S markierte Cyclin A und B Konstrukte mit und ohne den Proteasomen-Inhibitor N-Ac-Leu-Leu-norleucinal getestet. A: Das Cyclin A ist nach 45 Minuten vollständig abgebaut. Nach 15 Minuten erscheint die höhermolekulare Modifikationsbande. Durch die Zugabe von N-Ac-Leu-Leu-norleucinal wird der Abbau von Cyclin A verhindert, aber auch hier erscheint die höhermolekulare Bande nach 15 Minuten. B: Das Cyclin A∆169 (Cyclin Ad169) wird nicht abgebaut. Nach 15 Minuten ist eine höhermolekulare Bande sichtbar. Diese Modifikation wird durch N-AcLeu-Leu-norleucinal verhindert. C: Das N-terminale 169 Aminosäuren lange Konstrukt von Cyclin A wird über den gemessenen Zeitraum von 70 Minuten kontinuierlich abgebaut. Durch Zugabe von N-Ac-Leu-Leunorleucinal wird der Abbau verhindert. Eine höhermolekulare Bande kann nicht beobachtet werden. D: Das Cyclin B wird über den gemessenen Zeitraum von 70 Minuten kontinuierlich abgebaut. Durch die Zugabe von N-Ac-Leu-Leu-norleucinal wird der Abbau verhindert. Eine höhermolekulare Bande kann nicht nachgewiesen werden. E: Das Cyclin B∆46 (Cyclin Bd46) verhält sich wie das Cyclin B. 30 3.1.5. Caspase-Inhibitoren Die Caspasen bilden eine eigenständige Familie der Proteasen, sog. cysteinyl aspartate-specific proteinases. (Nicholson, 1997). Grundsätzlich handelt es sich bei ihnen um Cystein Proteasen, die spezifisch nach einem Aspartat-Rest schneiden. Im Interphase-Extrakt von Xenopus wurde gezeigt, daß auch Cyclin A ein Zielprotein für Caspasen darstellt und während der EGT (early gastrulation transition) von einer ICE ähnlichen Caspase abgebaut wird. Der EGT stellt einen kritischen Punkt in der Embryonalentwicklung von Xenopus dar. Hier wird von der maternalen Transkription auf die zygotische Transkription umgestellt. Davon betroffen ist auch die Transkription von Caspase-Inhibitoren. Fehlen diese Inhibitoren kommt es zur Apoptose. In dieser Zeit wird Cyclin A von der Caspase-3 abgebaut, einer Caspase die zur ICE Subfamilie gehört (Stack and Newport 1997). Der Abbau durch Caspasen ist sehr spezifisch und führt zur Fragmentierung der Zielproteine, wie auch bei den Cyclin A und B Konstrukten in den hier gezeigten Experimenten beobachtet wurde. Aufgrund dieser Beobachtung wurde untersucht ob die Stabilität von Cyclin A durch Caspase-Inhibitoren beeinflußt werden kann. Hierzu wurden die zwei Caspase-Inhibitoren benutzt, die im Xenopus-Extrakt zur Klärung der Cyclin A Instabilität eingesetzt wurden (Stack and Newport 1997). Die beiden Inhibitoren, Ac-YVAD-CHO (Caspase-1 Inhibitor) und Ac-DEVD-CHO (Caspase-3 Inhibitor), wurden im Embryonenextrakt mit HA-Cyclin A und HA-Cyclin A∆169 bei verschiedenen Konzentrationen getestet (Abb.11). Der Western-Blot zeigt eine nahezu komplette Inhibition des HA-Cyclin A Abbaus durch Ac-YVAD-CHO bei einer Konzentration von 1mM. Eine 100fach geringere Menge an Ac-YVAD-CHO genügte dagegen, um den Abbau von Cyclin A im Xenopus-Extrakt zu blockieren (Stack and Newport 1997). Für Ac-DEVD-CHO wurde bei einer Konzentration von 25µM eine fast vollständige Inhibition des Cyclin A Abbaus beobachtet. Auch hier liegt die Konzentration 100fach über der in der Literatur bei Xenopus erzielten Inhibition des Cyclin A Abbaus (Stack and Newport 1997). Da das HA-Cyclin A∆169 im Embryonenextrakt stabil bleibt, scheinen nur die ersten 169 Aminosäuren von Cyclin A für den Abbau entscheidend zu sein. Die N-Terminalen 169 Aminosäuren wurden auf das Vorhandensein von Caspase Erkennungssequenzen hin analysiert. Die Konsensussequenz der meisten Caspasen DXXD, konnte im N-Terminus von Cyclin A nicht gefunden werden. Die hohe Konzentration der Caspase-Inhibitoren, die eingesetzt werden mußte, um eine Inhibition des Cyclin A Abbaus zu erreichen, schließt die Beteiligung von Caspasen beim Abbau von Cyclin A und B aus. Durch 31 die hohe Konzentration des Inhibitors können auch andere Proteasen inhibiert worden sein. Die Identität dieser Proteasen wurde nicht weiter untersucht. Abb. 11: Titration des Caspase 1 Inhibitors Ac-YVAD-CHO und des Caspase 3 Inhibitors Ac-DEVDCHO. Wie auch bei der Titration des Proteasomen-Inhibitors, wurde auch hier nur der 0 Minutenwert als Kontrolle für die eingesetzte Proteinmenge gewählt. Der 45 Minutenwert wurde genutzt, als der Zeitpunkt, bei dem Cyclin A im Extrakt ohne Inhibitoren abgebaut wird. Die Kontrollen (Ko.) erstrecken sich über beide Diagramme. Bei diesen Kontrollen wurde kein Inhibitor zugesetzt. Zu sehen ist hierbei, daß das HACyclin A nach 45 Minuten vollständig abgebaut ist, während HA-Cyclin A∆169 (HA-Cyclin Ad169) weitgehend stabil ist. A: Der Abbau von HA-Cyclin A wird bei einer Konzentration von 1mM AC-YVADCHO vollständig inhibiert. B: Durch Zugabe von 25µM AC-DEVD-CHO wird der Abbau von HA-Cyclin A verhindert. 32 3.1.6. Der Einfluß des Alters bzw. des Entwicklungsstadiums der Embryonen auf die Stabilität von Cyclin A In den bisherigen Experimenten wurde ein Extrakt aus 0-1 Stunde alten Embryonen benutzt. Die Embryonen dieses Alters befinden sich in den ersten 7 Teilungszyklen. Bei diesen Zyklen handelt es sich um sehr schnelle Kernteilungen, die nur ca. 8,6 Minuten dauern. In diesen Kernzyklen ist Cyclin A in vivo instabil. Nach den schnellen Zellzyklen verbringen die Zellen die meiste Zeit des Zellzykluses in der G2-Phase, in welcher Cyclin A stabil ist. Diese Bedingungen findet man in 2-4 Stunden alten Embryonen vor. Im 8-9 Stunden altem Extrakt befinden sich die meisten Zellen in der G1-Phase, in welcher Cyclin A instabil ist. In den nachfolgenden Experimenten soll gezeigt werden, ob der in vitro beobachtete Abbau der CyclinKonstrukte abhängig ist vom Alter der Embryonen. Das Cyclin A und das stabile Cyclin A∆169 zeigen in den 0-1 Stunde alten Embryonen das bereits bekannte Bild. Cyclin A ist nach 70 Minuten abgebaut und zeigt die bekannten Banden während Cyclin A∆169 stabil bleibt. In 2-4 Stunden alten Embryonen ist Cyclin A stabil. Sowohl das Cyclin A wie auch das Cyclin A∆169 zeigen nach 70 Minuten die bekannte höhermolekulare Isoformbande, aber nur einen geringen Abbau von Cyclin A. In dem noch älteren 8-9 Stunden alten Embryonenextrakt wird Cyclin A wiederum abgebaut während das Cyclin A∆169 stabil bleibt. Vergleicht man das durch Cyclin A Abbau erzeugte Bandenmuster zwischen dem 0-1 Stunden und dem 8-9 Stunden Extrakt, zeigt sich bei dem älteren Extrakt ein deutlich stärkerer Abbau (Abb.12). Durch den Einsatz von 2,4mg/ml N-Ac-Leu-Leu-norleucinal wird der Cyclin A Abbau in allen drei Extrakten verhindert (Abb.13). Wie schon in vorherigen Versuchen gezeigt, erscheint auch hier eine Bande nach 70 Minuten, die eigentlich für einen partiellen Cyclin A Abbau spricht. Banden, die für eine Ubiquitinierung von Cyclin A durch einen aktiven APC sprechen würden, treten auch beim 8-9 Stunden alten Extrakt nicht auf. Dies könnte bedeuten, daß der Cyclin A Abbau im 8-9 Stunden Extrakt ebenfalls unabhängig vom APC erfolgt oder daß es in diesem Versuchsansatz nicht möglich ist, eine Polyubiquitinierung nachzuweisen. Bei den älteren Extrakten ist auffallend, daß durch N-Ac-Leu-Leu-norleucinal eine Modifizierung des Cyclin A∆169 ausbleibt. Dies könnte bedeuten, daß N-Ac-Leu-Leu-norleucinal ein anderes Protein inhibiert wird, welches einen direkten oder indirekten Einfluß auf die Modifikation von Cyclin A∆169 haben könnte. 33 Abb. 12: Stabilität von Cyclin A und Cyclin A∆169 in unterschiedlich altem Embryonenextrakt. Die Stabilität der Cyclin A Konstrukte in 0-1 Stunden, 2-4 Stunden und 8-9 Stunden altem Embryonenextrakt. Entnommen wurden die Proben nach 0 Minuten, um die eingesetzte Ausgangsproteinmenge mit dem erwarteten Abbau nach 70 Minuten vergleichen zu können. Das Cyclin A ∆169 ist in allen drei Extrakten stabil und zeigt nach 70 Minuten eine höhermolekulare Bande. Das Cyclin A ist nur in 2-4 Stunden altem Extrakt stabil. Im 0-1 und 8-9 Stunden altem Extrakt wird Cyclin A abgebaut. Abb. 13: Einfluß des Proteasomen-Inhibitors N-Ac-Leu-Leu-norleucin auf den Abbau von Cyclin A und Cyclin A∆169 in unterschiedlich altem Embryonenextrakt. Stabilität der Cyclin A Konstrukte in 0-1 Stunden, 2-4 Stunden und 8-9 Stunden altem Embryonenextrakt. Entnommen wurden die Proben nach 0 Minuten, um die eingesetzte Ausgangsproteinmenge mit dem erwarteten Abbau nach 70 Minuten vergleichen zu können. Cyclin A und Cyclin A∆169 sind in allen drei Extrakten stabil. 34 Die Stabilität von endogenem Cyclin A und Cyclin B Bisher wurde die Stabilität von in vitro transkribierten und translatierten Cyclin A und Cyclin B Konstrukten in Embryonenextrakten getestet. Um zu testen wie stabil das endogene Cyclin A und Cyclin B in den verschiedenen Extrakten ist, wurden die Extrakte mittels Western-Blot analysiert. Das Experiment wurde unter den gleichen Bedingungen durchgeführt wie die Experimente, die zuvor durchgeführt wurden. Der Proteinextrakt, gewonnen aus 0-1 Stunden alten Embryonen, entstammt den Kernteilungsstadien 2 bis 7. Untersuchungen haben ergeben, daß die Menge an Cyclin A und B im Präblastodermstadium konstant ist. Behandelt man die Embryonen mit Cyclohexamid und verhindert dadurch die Proteinsynthese, ist schon nach 10 Minuten kein Cyclin A mehr nachweisbar, während Cyclin B über den gemessenen Zeitraum von 40 Minuten stabil bleibt (Edgar, et al., 1994). In zwei Stunden alten Embryonen hat die Zellularisierung bereits statt gefunden. Nach der Zellularisierung wird die Interphase durch das Auftreten der G2-Phase deutlich länger und die zygotische Genexpression beginnt. Im zellulären Blastodermstadium nimmt das Plasmavolumen pro Zellkern deutlich ab. Dazu kommt noch, daß die Zellteilungen nicht mehr synchronisiert verlaufen. Infolgedessen sind nicht in jeder Zelle Cyclin A und B vorhanden. In den 8-9 Stunden alten Embryonen befinden sich die meisten Zellen in der G0-Phase (Ruhephase). Jeder dieser Embryonenextrakte verfügt über eine unterschiedliche Proteinzusammensetzung. Welchen Einfluß die jeweilige Proteinzusammensetzung auf die Stabilität von endogenem Cyclin A und B hat, soll in den beiden folgenden Experimenten gezeigt werden. Das endogene Cyclin A wird in 0-1 Stunden altem Extrakt abgebaut (Abb. 14 A). Der Abbau des endogenen Cyclin A verläuft im Vergleich zum Abbau des in vitro translatierten Cyclin A (Abb.4 A) deutlich langsamer. Es sind auch mehrere Isoformen des Cyclin A vorhanden, wobei die Bande mit der geringsten Laufgeschwindigkeit am stärksten abgebaut wird. Im 2-4 Stunden altem Extrakt wird nur sehr wenig Cyclin A abgebaut. Aber auch hier sind mehrere Cyclin A Isoform-Banden zu erkennen (Abb.14 B). In den 8-9 Stunden alten Extrakten ist insgesamt weniger endogenes Cyclin A vorhanden. Das Cyclin A wird aber auch hier abgebaut (Abb.14 C). In allen drei Extrakten befindet sich ein ca. 40 kDa großes Protein das von dem hier benutzten Anti-Cyclin A Antikörper erkannt wird. Dieses zusäztliche Protein bleibt über den gemessenen Zeitraum hinweg stabil. 35 Abb. 14: Stabilität von endogenem Cyclin A. Aus dem bei 25°C inkubierten Embryonenextrakt wurden nach den angegebenen Zeitintervallen Proben entnommen und über ein 10%iges SDS-PAGE wurden die Proteine getrennt. Das endogene Cyclin A wurde mittels anti-Cyclin A Antikörpern im Western-Blot nachgewiesen. A: Extrakt aus 0-1 Stunden alten Embryonen. Cyclin A wird abgebaut, wobei der stärkste Abbau bei der Bande mit geringster Laufgeschwindigkeit beobachtet werden kann. B: Extrakt aus 2-4 Stunden alten Embryonen. Der Cyclin A Abbau ist sehr gering. C: Extrakt aus 8-9 Stunden alten Embryonen. Es ist weniger endogenes Cyclin A vorhanden. Das vorhandene Cyclin A wird aber kontinuierlich abgebaut. 36 Das endogene Cyclin B verhält sich ähnlich wie das endogene Cyclin A. Das heißt es wird im 0-1 und auch im 8-9 Stunden altem Extrakt abgebaut während es im 2-4 Stunden altem Extrakt relativ stabil bleibt. (Abb.15 A, B und C). Abb. 15: Stabilität von endogenem Cyclin B. Aus dem bei 25°C inkubierten Embryonenextrakt wurden nach den angegebenen Zeitintervallen Proben entnommen und über ein 10%iges SDS-PAGE wurden die Proteine getrennt. Das endogene Cyclin B wurde mittels anti-Cyclin B Antikörpern im Western-Blot nachgewiesen. A: Extrakt aus 0-1 Stunden alten Embryonen. B: Extrakt aus 2-4 Stunden alten Embryonen. C: Extrakt aus 2-4 Stunden alten Embryonen. 37 3.2. Keimbahntransformation der HA-Cyclin A und HA-Cyclin A∆169 Gene Wie die Experimente zur Stabilität von Cyclin A Konstrukten in Embryonenextrakten gezeigt haben, verhält sich das Cyclin A ∆169 im Gegensatz zum Cyclin A stabil. Um zu untersuchen ob die Instabilität von Cyclin A für einen normalen Zellzyklus, vor allem für die Aufrechterhaltung der Interphase verantwortlich ist, wurden transgene Fliegen erzeugt, die zusätzlich zu maternalem Wildtyp Cyclin A, maternale Cyclin A∆169 mRNA im Embryo exprimieren. Zu diesem Zweck wurden transgene Fliegen hergestellt, die das HA-Cyclin A und das HA-Cyclin A∆169 maternal exprimieren sollen. Die Translation der mRNA im Ei soll hierbei erst nach der Befruchtung und der Eiablage erfolgen. Eine maternale Expression von HA-Cyclin A∆169 könnte den Zellzyklus in den sich entwickelnden Ovarien stören und die Bildung von Eiern verhindern. Eine maternale Expression und Translation der mRNA erst nach der Eiablage wird z.B. unter nanos Kontrolle beobachtet (Lehmann, and Nüsslein-Volhard, 1991). Beim nanos Promotor und nanos Translationssignal sind die hierfür notwendigen Sequenzen bekannt und werden durch ein ca. 900bp großes Promotor-Fragment und ein ca. 800bp langes 5’UTR gegeben. Zusätzlich wurde die Lokalisierungssequenz des bicoid-Gens verwendet (Ferrandon and Nüsslein-Vollhard, 1986). Diese Sequenz vermittelt die Lokalisation der mRNA am anterioren Pol des Embryos. Die Kombination aus bicoid Lokalisation, nanos-Promotor und Translationssignal soll dazu dienen, daß das Cyclin A Konstrukt maternal exprimiert, die mRNA nur am anterioren Pol lokalisiert und erst nach der Eiablage translatiert wird. Dadurch sollte sich ein Cyclin A Proteingradient im Embryo ausbilden. Infolgedessen wäre es möglich, abhängig von der Position des Gradienten entlang der anterior-posterior Achse verschiedene HA-Cyclin A und des HA-Cyclin A∆169 Konzentration zu testen. 38 ∆169 3.2.1. Klonierung der zu injizierenden HA-Cyclin A und HA-Cyclin A Konstrukte zur Erzeugung transgener Fliegen Die für das HA-Cyclin A und HA-Cyclin A∆169 kodierenden Abschnitte wurden aus dem pSP64 Vektor upstream vom nanos-Promotor in den Bluescript SK(-)-Vektor subkloniert (Abb.16). Abb. 16: Struktur der nos-HA Cyclin A und nos-HA Cyclin A∆169 Vektoren. T7 und T3: Promotor für die T7 und die T3 Polymerase. Die Plasmide Bluescript SK(-) enthalten einen ca. 900bp großen nanos-Promotor am 5’-Ende, dem folgt ein ca. 20bp großes HA-tag. Im Anschluß an das HA-tag befindet sich das ca. 1600bp große Cyclin A (A) bzw. das ca. 1000bp große HA-Cyclin A∆169 (B). Das am 3´-Ende des bicoid lokalisierte Lokalisationssignal wurde, nach Entfernung aus dem Bluescript SK(-)-Vektor, mit den gleichfalls subklonierten nanos-Promotor-tragenden HA Cyclin A und HA Cyclin A∆169 Abschnitten verknüpft (Abb.17). Abb. 17: Struktur der 5’nos-HA Cyclin A-3’bcd und 5’nos-HA Cyclin A∆169-3’bcd Vektoren. T7 und T3: Promotor für die T7 und die T3 Polymerase. Die Plasmide Bluescript SK(-) enthalten ein ca. 900bp großen nanos-Promotor am 5’-Ende, dem folgt ein ca. 20bp großes HA-tag. Im Anschluß an das HA-tag befindet sich das ca. 1600bp große Cyclin A (A) bzw. das ca. 1000bp große HA-Cyclin A∆169 (B). Das bcd 3’ UTR (bicoid-Sequenz) hat eine Länge von ca. 700bp. 39 Diese unter der nanos/bicoid Kontrolle stehenden Cyclin A Konstrukte wurden schließlich in den pW8 Vektor kloniert (Abb.18). Der pW8 Vektor enthält ein white Genkonstrukt. Dieses Konstrukt kodiert für ein Gen, welches im white- Hintergrund zu einem roten Augen-Phänotyp führt. Der auf diese Weise entstandene pW8 Vektor wurde nun zusammen mit dem Transposase-tragenden Helferplasmid P∆2-3 in white-- Embryonen injiziert (siehe Methoden 2.2.3.). Abb. 18: Struktur der 5’nos-HA Cyclin A-3’bcd und 5’nos-HA Cyclin A∆169-3’bcd Vektoren. Die pW8 Plasmide enthalten ein white-Genkonstrukt welches für ein Gen kodiert, daß für den roten Augen-Phänotyp verantwortlich ist. Am 5’-Ende befindet sich ein ca. 900bp großer nanos-Promotor, dem folgt ein ca. 20bp großes HA-tag. Im Anschluß an das HA-tag befindet sich das ca. 1600bp große Cyclin A (A) bzw. das ca. 1000bp große HA-Cyclin A∆169 (B). Das bcd 3’ UTR (bicoid-Sequenz) hat eine Länge von ca. 700bp. 3.2.2. Erzeugung transgener Fliegen Die zuvor beschriebenen 5’nos-HA Cyclin A-3’bcd und 5’nos-HA Cyclin A∆169 -3’bcd pW8 Vektoren wurden zusammen mit dem Hilfsvektor P∆2-3 in ca. 300 white-Embryonen injiziert (siehe Methoden 2.2.3.). Nach ca. 24 Stunden entwickelten sich bei Raumtemperatur die Larven. Aus den 300 mit 5’nos-HA Cyclin A-3’bcd injizierten Embryonen entwickelten sich 20 Larven. Aus den 300 mit dem 5’nos-HA Cyclin A∆169 -3’bcd injizierten Embryonen 32 Larven. Alle diese Larven entwickelten sich zu adulten Fliegen, die 40 - einzeln mit white Jungfrauen bzw. Männchen gekreuzt wurden. Die Nachkommen dieser Kreuzung sollten den roten Augenphänotyp zeigen, wenn sie das Transgen tragen. Die Einzelkreuzungen ergaben für das 5’nos-HA Cyclin A-3’bcd 5 unabhängige transgene Linien. Dabei wurden insgesamt 35 transgene Männchen und Weibchen im fast ausgeglichenen Geschlechterverhältnis erhalten. Die Einzelkreuzungen für das 5’nos-HA Cyclin A∆169 -3’bcd ergaben nur 3 unabhängige transgene Linien mit insgesamt 38 transgenen Weibchen und keinem Männchen. Erwartet wurde eine normale Geschlechterverteilung. Wenn das Cyclin A∆169 einen Einfluß auf den Zellzyklus hat, sollten die Weibchen allerdings steril sein und die Embryonalentwicklung gestört werden. 3.2.3. Nachweis von HA-Cyclin A und HA-Cyclin A∆169 in transgenen Embryonen Für das Auftreten normal entwickelter Nachkommen aus den transgenen Weibchen gibt es zwei Erklärungen. Das HA-Cyclin A∆169 könnte keinen Effekt auf den Zellzyklusverlauf haben, oder das HACyclin A∆169 wurde in zu geringer Menge exprimiert. Zum Nachweis der in den transgenen Embryonen exprimierten HA Cyclin A Proteine wurden daher 5’nosHA Cyclin A∆169 -3’bcd und 5’nos-HA Cyclin A-3’bcd transgene Weibchen mit white--Männchen gekreuzt und eine ausreichende Anzahl von Embryonen für einen Proteinextrakt gesammelt. Dieses Proteinextrakt wurde auf ein Polyacrylamidgel aufgetragen und mit HA-Antikörper entwickelt (Abb. 19). Leider konnte weder HA-Cyclin A noch HA-Cyclin A∆169 in den Embryonen nachgewiesen werden. Zu diesem Zeitpunkt muß es daher offen bleiben ob die untersuchten Cyclin A Transgene in funktioneller Form exprimiert werden können. (siehe 4.). 41 Abb. 19: Western Blot zum Nachweis von HA-Cyclin A und HA-Cyclin A∆169 in den Embryonen transgener Fliegen. Es wurde ein Proteinextrakt aus 20 Embryonen auf ein Polyacrylamidgel aufgetragen. Als Kontrolle wurde in vitro translatiertes HA-Cyclin A und HA-Cyclin A∆169 aufgetragen. 3.3. Injektion von RNA in Drosophila Embryonen 3.3.1. Testung der Transkriptionsbedingung Da die Keimbahntransformation nicht den gewünschten Erfolg brachten, wurde eine weitere Methode angewendet, um Proteine in frühen Embryonen zu exprimieren. Die Cyclin A Konstrukte wurden in Form von RNA in die frühen Embryonen injiziert. Hierzu wurde RNA in vitro transkribiert (2.2.4.). Für eine erfolgreiche Expression von Proteinen ist die Qualität der RNA entscheidend. Die Qualität der RNA kann durch in vitro Translation (2.2.1.4.) bestimmt werden (Abb.20 A). Es hat sich jedoch gezeigt, daß eine erfolgreiche in vitro Translation nichts darüber aussagt, wie gut sich die RNA in vivo translatieren läßt. Die geeigneten Transkriptionsbedingung für eine erfogreiche in vivo Translation wurden mit Hilfe des WesternBlots analysiert. Um die durch RNA Injektion exprimierten Cyclin A Proteine von dem endogenen Cyclin A unterscheiden zu können, wurden zunächst Cyclin A Konstrukte mit einem HA-tag benutzt. Die RNA-Injektion erfolgte in 0-1 Stunden alten Embryonen. Die Embryonen wurden dechorionisiert (2.2.3.4.), auf einem Deckglas aufgereiht und getrocknet (2.2.3.5.). Es wurde eine 1,5mg/ml RNA Lösung injiziert (2.2.3.5.). Nach Injektion erfolgte eine Inkubation von 45 Minuten bei Raumtemperatur. Anschließend wurden die Embryonen in Laemmli-Puffer homogenisiert, die Proteine über ein 10%iges PAGE aufgetrennt und mit Hilfe von verschiedenen Antikörpern, entsprechend den injizierten RNAs, im Western-Blot nachgewiesen. 42 Zur Ermittlung der geeigneten Transkriptionsbedingungen, die eine erfolgreiche in vivo Translation ermöglicht, wurde linearisiertes und nicht linearisiertes Cyclin A∆169 und HA- Cyclin A∆169 in vitro mit Hilfe der SP6 Polymerase transkribiert und die RNA in Embryonen injiziert. Die in vitro Transkription von HACyclin A∆169 erfolgte nach Linearisierung einmal mit Cap (m7G(5’)ppp(5’)G) und einmal ohne Cap. Wie in Abb.21 B deutlich zu sehen ist, war es zur erfolgreichen Durchführung der Experimente entscheidend, daß die Plasmide zur in vitro Transkription linearisiert werden (Abb.20 B, Spur 2, 3 und 6) und die synthetisierten RNAs ein Cap (m 7G(5’)ppp(5’)G) trugen. Bleibt das Linearisieren des Plasmids vor der in vitro Transkription aus, wird nur sehr wenig Protein im Embryo gebildet (Abb.20 B, Spur 4). Fehlt das Cap im in vitro Transkriptionsansatz, wird im Embryo kein Protein von der injizierten RNA gebildet (Abb.20 B, Spur 5). Abb. 20: Überprüfung der in vivo Translatierbarkeit unterschiedlicher in vitro transkribierter mRNAs Die in vitro Transkription der beiden Cyclin A Konstrukte erfolgte durch die SP6 Polymerase. Der Promotor für die SP6 Polymerase befindet sich im SP6 Vektor vor der Klonierungsstelle. Alle Konstrukte besitzen ein 3’ β-globin Gen aus Xenopus. Dieses β-globin Gen erhöht die Effizienz der Translation. Das HA-Cyclin A∆169 enthält zusätzlich noch einen poly-A Schwanz mit 20 As. In Xenopus wird durch die 3’Region und mit Hilfe des poly-A Schwanzes sowohl die Translationseffizienz wie auch die Stabilität der RNA erhöht. Die Konstrukte wurden zum Vergleich der besseren in vivo Translatierbarkeit, einmal mit EcoRI linearisiert oder ungeschnitten transkribiert. Die RNA wurde in 1 Stunde alte Embryonen injiziert und anschließend 45 Minuten inkubiert. Die in vivo translatierten Proteine wurden auf einem Polyacrylamidgel aufgetrennt und der Western-Blot mit dem affinitätsgereinigten Anti-Cyclin A Antikörpen entwickelt. A: In vitro Translationkontrolle. Die Qualität der RNA scheint keinen Einfluß auf die Translation in vitro zu haben. B: Die Transkriptionsbedingungen spielen bei der in vivo Translation eine große Rolle. Die RNA von zuvor linearisierte Plasmide lassen sich in vivo besser translatieren (Spur 2 und 3 und 6). Wurde das Plasmid vor der Transkription nicht linearisiert, erhält man eine geringere Proteinmenge (Spur 4). Fehlt dagegen das Cap im Transkriptionsansatz, wird die in Embryonen injizierte RNA nicht translatiert. 43 3.3.2. Einfluß der in vivo translatierten Cyclin A Konstrukte auf den Zellzyklus Wie in dem vorhergehenden Western-Blot (Abb. 20 B) zu sehen ist, wurde die in vitro transkribierte RNA der Cyclin A Konstrukte in den 1 Stunde alten Embryonen translatiert, wenn linearisierte Plasmide zur Transkription eingesetzt wurden und die RNA ein Cap besitzt. Durch Expression der Cyclin A Konstrukte in 1 Stunde alten Embryonen soll der Einfluß der Konstrukte auf den Zellzyklusverlauf untersucht werden. Die mit RNA injizierten Embryonen wurden 45 Minuten bei Raumtemperatur inkubiert, anschließend fixiert und detvitellinisiert (siehe 2.2.4.2). Die Entfernung der Vitellinmembran ist notwendig, um Färbungen wie z.B. der DNA oder Antikörper-Färbungen durchzuführen. Durch die Injektion von HA-Cyclin A, HA-Cyclin AR160G und HA-Cyclin A∆169 RNA war es darüberhinaus auch möglich, deren Translation in vivo durch AntiHA Antikörper nachzuweisen. Als weiterer Zellzyklusmarker wurden die phosphorylierten H3 Histone mit Hilfe von PH3 Antikörpern nachgewiesen (Su, et al., 1998). Phosphorylierte H3 Histone findet man normalerweise nur von der Prophase bis zur Telophase, wenn die Chromosomen kondensiert sind. Die Kombination der DNA-Färbung mit der Antikörperfärbung der HA-Cycline ermöglicht es, die beobachteten Einflüsse auf den Zellzyklus mit dem injizierten Cyclin in Verbindung zu bringen. Als Kontrolle wurde zunächst Wasser injiziert, um durch die Injektion bedingte mechanische Effekte auf den Zellzyklusverlauf zu beobachten. Durch die Injektion von Wasser wurden keine Einflüsse auf den Zellzyklus nachgewiesen. Mehr als 90% der Embryonen entwickelten sich normal. Die restlichen Embryonen waren entweder nicht befruchtet oder wiesen so starke mechanische Schäden auf, daß sie sich nach der Injektion nicht mehr weiter entwickeln konnten. Als weitere Kontrolle wurde die RNA eines Proteins injiziert, welches keinen Einfluß auf den Zellzyklus hat. Hierfür wurde die RNA benutzt, die für einen Teil des Torso-Proteins codiert. Torso ist ein Transmembranprotein, was keinen Einfluß auf den Zellzyklus zeigt (Sprenger F., 1992). Das hier verwendete Torso Konstrukt (TU025) codiert nur für den cytoplasmatischen Teil des Transmembranproteins. Zunächst wurde wieder überprüft ob die Torso RNA in vivo translatiert wird. Hierzu wurde die in vitro transkribierte Torso RNA (1,5mg/ml) in 1 Stunde alte Embryonen injiziert. Nach 45 Minuten wurden die Embryonen in Lämmli homogenisiert und die Proteine über ein SDS-PAGE aufgetrennt. Mit Hilfe spezifischer Anti-Torso Antikörper konnte das Torso-Protein im Western-Blot nachgewiesen wurden (Abb. 21). Die Spuren 2-5 zeigen eine höhermolekulare schwache Bande, die das endogende Torso darstellt. Bei der höhermobilen, stärker ausgeprägten Bande handelt es sich um das injizierte TU25-torCy Konstrukt. In Spur 1 ist zum Verglich in vitro translatiertes Torso-Konstrukt aufgetragen. Als Kontrolle für die 44 ∆169 Antikörperspezifität wurde Cyclin A RNA injiziert (Spur6). Hierbei wird nur endogenes Torso nachgewiesen. Abb. 21: Nachweis des in vivo translatierten torso Proteins nach mRNA Injektion. In 0-1 Stunden alte Wiltyp-Embryonen wurde in vitro synthetisierte torso mRNA (1,5mg/ml) injiziert. Nach der Injektion wurden die Embryonen 45 Minuten bei Raumtemperatur inkubiert und anschließend wurde ein Proteinextrakt hergestellt. Die aufgetragene Proteinmenge pro Spur entspricht 10 aufgearbeiteten Embryonen. Diese Proteine wurden über ein 10%iges SDS-PAGE aufgetrennt und mit Hilfe von αtorso Antikörpern im Western-Blot entwickelt. Spur 2-5 zeigt eine höhermolekulare Bande. Hierbei handelt es sich um das endogene Torso und eine zweite niedermolekulare Bande, die dem injizierten Torso Konstrukt entspricht. Spur 1 zeigt in vitro translatiertes Torso-Konstrukt (TU025). Als Kontrolle wurde Cyclin A∆169 RNA injiziert (Spur 6). Hier ist nur das endogene Torso zu sehen. 45 Es konnte also gezeigt werden, daß in vitro transkribierte torso RNA in vivo translatiert werden kann. Diese RNA wurde anschließend in 0-1 Stunden alte Embryonen injiziert, für 45 Minuten inkubiert und die DNA mit Propidiumjodid (PPI) angefärbt. Die Abb.22 zeigt einen repräsentativen Embryo, der sich nach der Injektion von torso RNA normal entwickelt hat und keinen Einfluß auf den Zellzyklus erkennen läßt. Von den 61 injizierten Embryonen haben sich ungefähr 83% normal entwickelt, ca. 3% der Eier waren unbefruchtet und 14% der Embryonen zeigten Injektionsartefakte. Abb. 22: Einfluß der injizierten torso RNA auf den Zellzyklus. Wildtyp-Embryo in der frühen Embryogenese (0-1 Std. alt , ca. dem 7. Zellzyklus entsprechend). Es wurde Torso RNA (1,5mg/ml) injiziert. Nach einer Inkubation von 45 Minuten wurden die Embryonen fixiert, detvitellinisiert und die DNA mit Propidiumjodid (PPI) gefärbt. Die DNA-Färbung zeigt einen sich normal entwickelten Embryo. Die Zellkerne sind gleichmäßig im Embryo verteilt und befinden sich alle in der Prophase. Als nächstes wurde RNA injiziert, die für ein normales Cyclin A codiert. Hierbei entwickelten sich von 53 injizierten Embryonen 76% normal, 11% waren unbefruchtet und 13% zeigten injektionsbedingte Artefakte. Die DNA Färbung mit Hoechstfarbstoff, zeigt gleichmäßig verteilte Zellkerne, die sich alle im gleichen Zellzyklusstadium befinden (Abb.23). 46 Abb. 23: Injektion in vitro transkribierter Cyclin A mRNA. Wildtyp-Embryo in der frühen Embryogenese (0-1 Std. alt , ca. dem 7. Zellzyklus entsprechend). Es wurde Cyclin A mRNA (1,5mg/ml) injiziert. Nach einer Inkubation von 45 Minuten wurden die Embryonen fixiert, detvitellinisiert und gefärbt. Die DNA-Färbung (Hoechst) zeigt einen sich normal entwickelten Embryo nach Cyclin A RNA Injektion. Die Zellkerne sind gleichmäßig im Embryo verteilt und befinden sich alle im gleichen Zellzyklus-Stadium. Die Torso Kontrolle hat gezeigt, daß durch die Injektion von RNA keine größeren Störungen des Zellzyklusverlauf hervorgerufen wurden. Durch die Injektion von normalem Cyclin A konnte gezeigt werden, daß eine erhöhte Menge an Cyclin A den Zellzyklus nicht stört. Bei der Injektion von RNA , die für das Cyclin A∆169 codiert, wurde jedoch der Verlauf des Zellzyklus massiv gestört. In diesem Experiment haben sich nur 15% der 71 injizierten Embryonen nach der Cyclin A∆169 RNA Injektion normal entwickelt, 7% waren unbefruchtet und 78% der Embryonen wiesen deutliche Effekte auf den Zellzyklusverlauf. Die Injektion der RNA erfolgte in Abb.24A in der Mitte des Embryos. In dem injizierten Bereich des Embryos sind weniger Kerne vorhanden. In der Mitte des Embryos wurden daher weniger Kerne angefärbt. An den beiden Enden des Embryos wurde die Proliferation nicht beeinträchtigt. Die gleiche Entwicklung ist in Abb.24C zu sehen. Hier wurde die RNA an einem Ende des Embryos injiziert. Die Färbung der phosphorylierten H3 Histone erfolgt normalerweise während der Mitose ,wenn die Chromosomen kondensieren. In dem nicht injiziert Bereich des Embryos wird keine PH3 Färbung sichtbar. Diese Kerne befinden sich daher in der Interphase. Dagegen zeigen die Chromosomen im injizierten Bereich eine Färbung mit PH3, d.h. in dem injizierten Bereich sind die Chromosomen kondensiert und die Kernmembran vermutlich aufgelöst (Abb.24B und D). 47 Abb. 24: Injektion in vitro transkribierter Cyclin A∆169 mRNA. Wildtyp-Embryo in der frühen Embryogenese (0-1 Std. alt , ca. dem 7. Zellzyklus entsprechend). Es wurde Cyclin A∆169 mRNA (1,5mg/ml) injiziert. Nach einer Inkubation von 45 Minuten wurden die Embryonen fixiert, detvitellinisiert und gefärbt. A: Die DNA wurde mit Hilfe von Propidiumjodid angefärbt. Es zeigt sich deutlich eine höhere Zellkerndichte in dem Bereich des Embryos in dem nicht injiziert wurde. In dem injizierten Bereich sind kondensierte Chromosomen zu sehen. B: Die phosphorylierten H3 Histone wurden durch Antikörperfärbung sichtbar gemacht. Diese Färbung zeigt, daß in dem Bereich, in dem die Zellkerne dichter sind, keine H3 Histone phosphoryliert sind, d.h. in diesem Bereich befinden sich keine Zellkerne in der Interphase. Im injizierten Abschnitt des Embryos weisen die Chromosomen phosphorylierte H3 Histone auf. Die bisherigen Resultate ließen Rückschlüsse bezüglich der Lokalisation des Cyclin A∆169 nur infolge der beobachteten Wirkung auf den Zellzyklus im injizierten Bereich zu. Die Injektion von HA-getagten Cyclin A RNA ermöglicht es, die von der injizierten RNA translatierten Proteine mit Hilfe von HA-Antikörper direkt nachzuweisen. In Abb.25 ist ein Embryo dargestellt bei dem die HA-Färbung ausgehend vom posterioren Pol ein Gradienten bildet, der das HA-Cyclin A bis zur Mitte hin detektierbar macht. Dies bedeutet, daß die injizierte RNA vermutlich am posterioren Ende des Embryos lokalisert wurde (Abb.25A). 48 Die DNA Färbung zeigt genau in dem Bereich, in welchem auch das HA-Cyclin A ∆169 Protein lokalisiert ist eine deutliche Reduktion der Proliferation (Abb. 25 B). Die Chromosomen sind in diesem Bereich kondensiert, während sich die Kerne im nicht injizierten Bereich alle in der frühen Interphase befinden (Abb.25E). Die PH3 Antikörperfärbung zeigt in dem Bereich in dem kein, oder nur wenig HA-Cyclin A∆169 vorhanden ist, keine Färbung. Die Kerne befinden sich demnach alle in der Interphase (Abb. 25 C und F). Dagegen zeigt die PH3 Färbung in dem Bereich mit höherer HA-Cyclin A∆169 Konzentration eine Phosphorylierung der H3 Histone und kondensierte Chromosomen. Die Betrachtung der Grenzzone zwischen injiziertem und nicht injizierten Bereich mit einer stärkeren Vergrößerung ermöglicht eine bessere Beurteilung, in welchem Stadium des Zellzykluses sich die Zellkerne befinden. Die DNA-Färbung zeigt, daß sich die Zellkerne im nicht injizierten Bereich alle in der frühen SPhase befinden. Das ist an den kleinen, homogen gefärbten Zellkernen gut zu erkennen (Abb.25D). In diesem Abschnitt des Embryos wird kein oder nur sehr wenig HA-Cyclin A∆169 nachgewiesen (Abb.25D). Ist dagegen viel HA-Cyclin A∆169 vorhanden, ist die Kernmembran aufgelöst und die Chromosomen sind kondensiert. Im Grenzbereich zwischen Interphase Kernen und kondensierten Chromosomen sind Chromosomen zu erkennen, die aus der Interphase heraus kondensieren (Abb. 25E und F). Betrachtet man sich die Lokalisation von HA-Cyclin A∆169 genauer, erkennt man eine überwiegende Lokalisation des HA-Cyclin A∆169 in den Kernen (Abb.25D). Dies ist nur im Grenzbereich möglich, da sich hier auch Interphase-Kerne befinden. Im Embryo mit hoher HA-Cyclin A∆169 Konzentration ist dagegen die Kernmembran aufgelöst. 49 Abb. 25: Injektion in vitro transkribierter HA-Cyclin A∆169 mRNA. Wildtyp-Embryo in der frühen Embryogenese (0-1 Std. alt , ca. dem 7. Zellzyklus entsprechend). Es wurde HA-Cyclin A∆169 mRNA (1,5mg/ml) injiziert. Nach einer Inkubation von 45 Minuten wurden die Embryonen fixiert, detvitellinisiert und gefärbt. A: Die HA-Antikörperfärbung zeigt den Teil des Embryos indem die injizierte mRNA translatiert wurde. B: Die DNA wurde mit Hilfe von Propidiumjodid angefärbt. Es zeigt sich deutlich eine höhere Zellkerndichte in dem Bereich des Embryos in dem nicht injiziert wurde. In dem injizierten Bereich sind kondensierte Chromosomen zu sehen. Die Zellkerne hier befinden sich vermutlich in der Metaphase. C: Die phosphorylierten H3 Histone wurden durch Antikörperfärbung sichtbar gemacht. Diese Färbung zeigt, daß in dem Bereich, in dem die Zellkerne dichter sind, sehr viele H3 Histone phosphoryliert sind, d.h. in diesem Bereich befinden sich die meisten Zellkerne in der S-Phase. Im injizierten Abschnitt des Embryos befinden sich die Zellen nicht in der S-Phase, die H3 Histone weisen aber trotzdem Phosphorylierung auf. D: Bei stärkerer Vergrößerung ist deutlicher zu sehen, daß sich die Kerne im injizierten Teil des Embryos nicht in der S-Phase befindet aber dennoch H3 Histone phosphoryliert sind. In dem Bereich des Embryos, der von der Injektion verschont blieb, sind die Kerne homogen und klein, dies spricht für die frühe S-Phase. Alle dieser Kerne zeigen eine PH3-Färbung. E: Der Nachweis der Proteinlokalisation mit Hilfe der HAAntikörpern zeigt bei stärkerer Vergrößerung, daß das HA-Cyclin A∆169 in den nicht injizierten Bereich diffundiert ist und sich dort hauptsächlich im Zellkern befindet. F: Die DNA-Färbung zeigt bei dieser Vergrößerung die lockeren Chromatinstrukturen, die sich aber schlecht einem bestimmten Stadium im Zellzyklus zuordnen lassen. Das Cyclin A∆169 zeigte deutliche Effekte auf den Verlauf des Zellzykluses. Ob diese Effekte auf das Fehlen der destruction box zurückzuführen sind, kann nicht ausgeschlossen werden. 50 Das Cyclin A Konstrukt mit der Punktmutation in der destruction box zeigte in den in vitro Stabilitätsexperimenten die gleiche Instabilität wie das normale Cyclin A. Durch RNA Injektion soll nun überprüft werden ob HA-Cyclin AR160G den Zellzyklusverlauf beeinflußt. Wird das eine Punktmutation tragende HA-Cyclin AR160G mRNA in die Embryonen injiziert ergibt sich das gleiche Bild wie bei der Injektion des normalen Cyclin A. Die DNA-Färbung ergibt einen gleichmäßigen Zellzyklusverlauf im gesamten Embryo (Abb. 26B und E). Durch die HA-Färbung kann das HA-Cyclin AR160G Protein an der injizierten Stelle, in der Mitte des Embryos, nachgewiesen werden (Abb.26A und D). Die PH3 Färbung zeigt, daß sich alle Kerne in der Interphase befinden (Abb.26C). Die Vergrößerung des Grenzbereiches zwischen injiziertem Abschnitt und nicht injiziertem Abschnitt zeigt, daß sich das HACyclin AR160G, im Unterschied zum HA-Cyclin A∆169, nicht im Zellkern befindet. Es liegt ausschließlich im Cytoplasma vor (Abb.26F). Womöglich sind infolge der cytoplasmatischen Lokalisation des Cyclin AR160G keine Effekte auf die Zellkernphasen zu beobachten. Die Injektion von Cyclin AR160G RNA ohne HA-tag ergab das gleiche Bild. Diese Resultate stehen im Einklang mit der Vermutung, daß das HA-tag keinen Einfluß auf die Lokalisation der Cyclin A Konstrukte hat. Abb. 26: Injektion in vitro transkribierter HA-Cyclin AR160G mRNA. Wildtyp-Embryo in der frühen Embryogenese (0-1 Std. alt , ca. dem 7. Zellzyklus entsprechend). Es wurde HA-Cyclin AR160G mRNA (1,5mg/ml) injiziert. Nach einer inkubation von 45 Minuten wurden die Embryonen fixiert, detvitellinisiert und gefärbt. A: Die DNA wurde mit Hilfe von Propidiumjodid angefärbt. Es zeigt sich hier eine weitgehend homogene Zellkerndichte im gesamten Embryo. Im Injektionsbereich haben die Zellkerne einen etwas größeren Abstand zueinander. B: Die HA-Antikörperfärbung zeigt den Teil des Embryos in welchem die injizierte mRNA translatiert wurde. C: Die phosphorylierten H3 Histone wurden durch Antikörperfärbung sichtbar gemacht. Diese Färbung zeigt, daß weitgehend alle Zellkerne phosphorylierte H3 Histone besitzen. D: Der Nachweis der Proteinlokalisation mit Hilfe der HA-Antikörpern zeigt bei stärkerer Vergrößerung, daß das HA-Cyclin AR160G in den nicht injizierten Bereich diffundiert ist und sich dort hauptsächlich im Cytoplasma befindet. 51 4. Diskussion Die ersten 13 Proliferationszyklen des Drosophila-Embryos sind synchrone Kernteilungen. Die Zyklen 2-9 bestehen nur aus der Mitose und der Replikation der DNA in der Interphase. In diesen ersten Zyklen tritt keine Fluktuation von Cyclinen auf ebenso fehlen signifikante Schwankungen der Cdc2-Kinaseaktivität. Aufgrund dieser Beobachtung ergaben sich eine Reihe von Fragen. Wie ist es möglich, in der Gegenwart von hoher Cdc2-Kinaseaktivität aus der Mitose in die Interphase zu gelangen? Wie wird die Interphase aufrecht erhalten und die DNA vollständig repliziert, bevor eine neue Mitose eingeleitet wird? Welche Mechanismen bestimmen den Zeitpunkt des Eintritts in die nächste Mitose? Welche Rolle dem Cyclin A in diesen ersten 2-9 Proliferationszyklen zukommt und wie die Stabilität des Cyclin A dessen Funktion beeinflußt wurde im Rahmen dieser Arbeit untersucht. 4.1. In vitro Untersuchungen zur Stabilität von Cyclin A Beide mitoserelevanten Cycline, Cyclin A und B, sind während der ersten 9 Zyklen in konstanter Menge vorhanden. Behandelt man Embryonen im Stadium 2-9 mit Cyclohexamid, wird die Proteinsynthese gehemmt und die Embryonen bleiben in der Interphase stecken. Dadurch kann bestimmt werden, welche Halbwertszeit die beiden Cycline haben. Bei diesem Versuch zeigte sich, daß Cyclin B bis zum Zyklus 8 relativ stabil ist. Das Cyclin A ist in diesen Versuchen jedoch schon nach 10 Minuten nicht mehr nachweisbar. Das heißt, Cyclin B ist während der ersten 8 Zyklen stabil während das Cyclin A abgebaut wird und ständig neu synthetisiert werden muß um seine konstante Konzentration im Embryo zu halten (Edgar B.A. et al, 1994). In der Interphase von Zyklus 14 sind dagegen beide Cycline stabil. Diese Instabilität von Cyclin A in den frühen Zellzyklen könnte ein Hinweis auf einen möglichen Regulationsmechanismus in den frühen Zyklen darstellen. In dieser Arbeit sollte untersucht werden, welche Mechanismen für die Instabilität verantwortlich sind und ob die Instabilität von Cyclin A für die Koordination dieser Zyklen von Bedeutung ist. Welcher Abbaumechanismus der Instabilität von Cyclin A zu Grunde liegt wurde in einem in vitro System, in dem Extrakt aus Embryonen der frühen Kernteilung benutzt wurden, untersucht. In diesem Extrakt wurde Cyclin A innerhalb von 15 Minuten abgebaut, während Cyclin A dem die Nterminalen 169 Aminosäuren (Cyclin A∆169) , einschließlich der destruction box fehlen, stabil bleibt. Die N-terminalen 169 Aminosäuren von Cyclin A wurden im Embryonen-Extrakt wie das normale Cyclin A kontinuierlich abgebaut. Diese Beobachtung legt die Vermutung nahe, daß die N-terminalen Aminosäuren, oder die in ihr lokalisierte destruction box, für die Stabilität von Cyclin A verantwortlich sind. 52 Aus diesem Grund wurde ein weiteres Cyclin A mit einer Punktmutation in der destruction box getestet (Cyclin AR160G). Dieses Konstrukt zeigte den gleichen Abbau wie das ungekürzte Cyclin A. Diese Punktmutation hat demnach keinen Einfluß auf die Stabilität des Cyclin A. Für Cyclin B konnte gezeigt werden, daß das stabile Cyclin B (Cyclin B∆46 ) in vivo stabil ist (Su, et al., 1998). In dem in vitro System wird jedoch sowohl Cyclin B wie auch Cyclin B∆46 abgebaut. Dieses Ergebnis zeigt, daß der Abbau von Cyclinen in dem in vitro System nicht destruction box abhängig erfolgt. Der destruction box abhängige Abbau wird durch Polyubiquitinierung eingeleitet. In dem in vitro System konnte bei Cyclin A eine höher molekulare Bande detektiert werden, deren Intensität durch Zugabe von Ubiquitin verstärkt wurde. Diese höher molekulare Bande fehlt bei dem Cyclin A mit der Punktmutation in der destruction box. Ob diese Bande durch Ubiquitinierung erfolgt konnte im Rahmen dieser Arbeit jedoch nicht untersucht werden. Um festzustellen, ob der Abbau durch die Aktivität von Proteasomen vermittelt wird, wurden zwei Inhibitoren eingesetzt, die die katalytische Aktivität der Proteasomen inhibieren. Unter diesen Bedingungen kann man bei Cyclinen, die destruction box abhängig abgebaut werden, die polyubiquitinierte Form der Cycline detektieren. In dem in vitro System konnte diese Polyubiquitinierung nicht beobachtet werden und nur einer der beiden eingesetzten Inhibitoren zeigte bei relativ hoher Konzentration eine Inhibition des Abbaus. Eine genauere Betrachtung des Abbaumusters von Cyclin A zeigt ein ca. 50 kDa großes Fragment, das in den verschiedenen Experimenten reproduzierbar nachgewiesen werden konnte. Bei einem Abbau durch Proteasomen entstehen dagegen nur kleine Peptide. Aufgrund dieser Fragmente kann geschlossen werden, daß der beobachtete Abbau von Cyclin A im Extrakt nicht destruction box abhängig erfolgt. In einem Interphase-Extrakt von Xenopus konnte gezeigt werden, daß Cyclin A nicht destruction box abhängig abgebaut wird, sondern durch die Caspase 3. Dieser Abbau wurde durch den Einsatz eines Caspase 3 Inhibitors verhindert (Stack and Newport, 1997). Auch hier wurde eine Fragmentierung von Cyclin A durch die Caspase 3 Aktivität beobachtet. In der vorliegenden Arbeit wurden zwei Caspase-Inhibitoren getestet, die Caspase 1 und 3 inhibieren. Durch diese Inhibitoren konnte eine nahezu vollständige Inhibition des Cyclin A Abbaus erzielt werden. Diese Inhibitoren mußten allerdings in 100fach höheren Konzentration als im Xenopus-Extrakt eingesetzt werden (Stack and Newport, 1997). Es ist also fraglich ob durch die hohe Konzentration des Inhibitors nicht unspezifisch andere Proteasen inhibiert worden sind. Da sich das Cyclin A∆169 im Extraktversuch stabil zeigt, scheinen sich die möglichen Schnittstellen für Proteasen in den ersten 169 Aminosäuren von Cyclin A zu befinden. In diesem Bereich von 53 Cyclin A gibt es jedoch keine Sequenz, die dem Konsensus (DXXD)der meisten bisher bekannten Caspasen entspricht. Eine Beteiligung von Caspasen am Abbau von Cyclin A ist daher unwahrscheinlich. Die Analyse der Proteolyse von Cyclin A im Extraktversuch wurde an diesem Punkt nicht weitergeführt. Um die Protease zu identifizieren wäre es notwendig gewesen eine Reihe weiterer Proteaseinhibitoren zu testen. Weiterhin könnte die Erkennungssequenz der Proteasen durch weitere verkürzte Cyclin A Konstrukten eingeengt werden. Die für diese Untersuchung benutzten Extrakte wurden aus Embryonen hergestellt, die sich in den frühen Kernzyklen befanden. In diesen Embryonen ist Cyclin A instabil, währen es in der 14. Interphase stabil ist. In Untersuchungen mit in vitro translatiertem Cyclin A in Extrakten aus 2-4 Stunden alten Embryonen ist das Cyclin A über den gemessenen Zeitraum von 70 Minuten stabil. Im Gegensatz dazu wird Cyclin A in Extrakten aus 8-9 Stunden alten Embryonen abgebaut. In diesem Extrakt befinden sich die meisten Zellen in der G1-Phase in der endogenes Cyclin A instabil ist. So kann eine Korelation von in vivo und in vitro Stabilität festgestellt werden. Es bedarf jedoch weiterer Experimente um zu ermitteln, ob es sich hierbei um die gleichen molekularen Mechanismen handelt. 4.2. Die Funktion der Stabilität von Cyclin A in den frühen Kernzyklen Cyclin A hat während der ersten Kernteilungen, sowohl in der Mitose wie auch in der Interphase, eine geringe Halbwertzeit. Die Instabilität während der Mitose ist wahrscheinlich auf den destruction box abhängigen Abbau durch den APC zurückzuführen. Die Instabilität von Cyclin A während der Interphase ist jedoch noch ungeklärt. Um zu testen ob diese Instabilität für einen normalen Zellzyklusverlauf wichtig ist, sollte das verkürzte Cyclin A in diesen Zyklen expremiert werden. Eine geeignete Methode für diese Untersuchung ist die Herstellung transgener Fliegenstämme. HA-Cyclin A und HA-Cyclin A∆169 sollten nach transgener Expression in vivo untersucht werden. Durch die Verknüpfung der Cyclin A kodierenden Genbereiche mit einer funktionellen nanos Promotor Sequenz und dem bicoid Lokalisationssignal sollte sichergestellt werden, daß die Cyclin A Proteine durch den nanos Promotor maternal und erst nach Befruchtung des Eis einen Gradienten im sich entwickelnden Embryo ausbilden. Danach sollte es möglich sein, die für die frühe Embryonalentwicklung relevanten biologischen Effekte von exprimierter Cyclin A Proteine genau zu untersuchen. In den transgenen Fliegen und den Nachkommen konnte jedoch kein HA-Cyclin A oder HA-Cyclin A∆169 nachgewiesen werden, und es wurde auch keine Zellzyklusabnormalität beobachtet. 54 Da diese Kombination von Cyclinen, nanos Promotor und bicoid Lokalisationssignal bisher noch nicht etabliert worden ist, besteht die Möglichkeit, daß entweder keine oder eine zu geringe Expression von Cyclin A erfolgte. Bei dem zweiten Ansatz wurde durch Injektion von in vitro transkribierter RNA eine Expression von Cyclin A erreicht. Durch Injektion von in vitro transkribierter RNA konnte in allen Fällen die gebildeten Proteine durch Western Blot oder durch Immunfärbungen nachgewiesen werden. In Kontrollversuchen wurde festgestellt, daß durch die Injektionen nur geringe Schäden verursacht wurden. So zeigten nach Injektion von normalem Cyclin A nur wenige Embryonen Defekte die meist auf die mechanische Beanspruchung der Embryonen zurück geführt werden konnten. Im Gegensatz dazu führte die Injektion von Cyclin A∆169 zu einer starken Störung des Zellzyklusverlaufs. Es wurde beobachtet, daß in dem injizierten Teil des Embryos die Proliferation der Kerne gestört wird. Die Chromosomen in diesem Bereich sind kondensiert und können mit Antikörpern gegen phosphoryliertes Histon 3 (PH3) angefärbt werden. Im Prinzip würde das für einen Arrest in der Mitose sprechen. Allerdings ist die Struktur der Chromosomen untypisch für ein Arrest, da keine für die Mitose typischen Strukturen erkannt werden können. Alternativ dazu könnte das verkürzte Cyclin A eine vorzeitige Kondensation der DNA aus der Interphase heraus verursacht haben. Diese Interpretation wird durch das Verhalten der Kerne im Grenzbereich zwischen dem gestörten und dem ungestörten Teil des Embryos unterstützt. Hier konnten direkt neben frühe Interphasekernen kondensierte Chromosomen entdeckt werden. Das heißt es entstand der Eindruck, daß die Interphasenkerne aufgelöst und die DNA kondensiert wurde. Der genaue Verlauf des Zellzyklus nach Injektion von stabilen Cyclin A sollte am besten in vivo beobachtet werden. Dies ist möglich in Embryonen, in denen die Chromosomen durch die Fusion von Histon H1 und GFP sichtbar gemacht werden können. Die RNA Injektion von Cyclin A mit einer Punktmutation in der destruction box (Cyclin AR160G) zeigte keinen Einfluß auf den Zellzyklusverlauf. Das Cyclin AR160G wird wie das Cyclin A im in vitro Stabilitätsversuch sehr schnell abgebaut. Wobei dieser Abbau unabhängig von der destruction box erfolgte. Interessant war die Beobachtung der Lokalisation der Cyclin A Konstrukte im Embryo. Während das Cyclin AR160G im Cytoplasma nachgewiesen wurde, befand sich das Cyclin A∆169 in den Interphasen-Zellkellkernen. In diesem Bereich wurde allerdings nur sehr wenig HA-Cyclin A∆169 nachgewiesen. In dem Teil des Embryos mit hoher HA-Cyclin A∆169 Konzentration waren die Chromosomen kondensiert und die Kernmembran aufgelöst. 55 Dies könnte darauf hindeuten, daß neben der Stabilität auch die subzellulare Lokalisierung von Bedeutung ist. Cyclin A ist in den Blastodermzellzyklen (ab Zyklus 14) während der Interphase im Cytoplasma lokalisiert. Während der Prophase befindet sich das Cyclin A im Zellkern. Ähnliches wurde für Cyclin B1 bei Eukaryoten beobachtet. Hierbei wurde eine Sequenz entdeckt, die für den Transport von Cyclin B1 aus dem Zellkern verantwortlich ist. Bei dieser Sequenz handelt es sich um ein nuclear export signal (Pines and Hunter, 1994). Dieses NES umfaßt etwa 50 Aminosäuren mit überdurchschnittlich vielen Leucinen. Diese Sequenz wurde in höhermolekularen Proteinen gefunden, die aus dem Zellkern ins Cytoplasma transportiert werden (Gorlich and Mattaj, 1996; Nigg, 1997). Das NES verursacht die cytoplasmatische Lokalisation von Cyclin B1 während des Zellzyklusverlaufs in der S- und G2-Phase. In der Prophase ist Cyclin B1 dagegen im Zellkern lokalisiert. Im Fall von Cyclin A ist bisher noch kein NES beschrieben worden. Da sich das Cyclin A∆169 im Gegensatz zum Cyclin A R160G im Zellkern befindet, könnte sich im N-terminalen Bereich von Cyclin A ein NES befinden. Da dem Cyclin A∆169 der N-Terminus fehlt, befindet sich dieses Konstrukt im Zellkern. Diese Lokalisierung im Kern könnte dazu führen, daß die Kernlamina durch Cyclin A ∆169 phosphoryliert werden kann, was dann schließlich zum Abbau der Kernmembran führt. Hierfür gibt es jedoch noch keine Beweise. Die durch das Cyclin A∆169 verursachte Blockade im Zellzyklus könnte demnach durch das NES vermittelt werden. Eine systematische Mutagenese des N-terminalen Cyclin A Bereiches ist notwendig, um eine der Funktion nach vergleichbare NES Sequenz zu identifizieren. Es konnte keine der Sequenz nach vergleichbare Leucin-reiche Sequenz in den 169 N-terminalen Aminosäuren in Cyclin A gefunden werden. Die Ergebnisse zeigen, daß eine Fehlregulation von Cyclin A den Zellzyklus stören können. Ob die beobachteten Effekte auf den Zellzyklus durch die Stabiltät des Cyclin A∆169 ,durch seine Lokalisation oder durch eine Kombination beider Beobachtungen zustande kommt, müßte noch genauer untersucht werden. 56 5. Zusammenfassung Die Regulation der frühen Kernteilungen in Drosophila ist noch weitgehend unverstanden, da keine Fluktuation von bekannten Zellzyklusproteinen beobachtet werden kann. Das Cyclin A zeigt in diesen Zyklen jedoch eine außergewöhnlich kurze Halbwertszeit. Die Funktion und Regulation des Cyclin A während der frühen Kernteilungen wurde in vitro und in vivo untersucht. Dazu wurde zuerst die Stabilität des Cyclin A in Embryonenextrakten analysiert. In diesem in vitro System konnte gezeigt werden, daß ein verkürztes Cyclin A (Cyclin A∆169), welchem die N-terminalen 169 Aminosäuren fehlen, im Gegensatz zum authentischen Cyclin A stabil ist. Im N-terminalen Bereich des Cyclin A befindet sich eine destruction box, die proteasomen-abhängig die proteolytische Degradation vermittelt. Der Abbau von Cyclin A in diesem System ist jedoch nicht destruction box abhängig und wird nur durch extrem hohe Konzentrationen an Proteaseinhibitoren gehemmt. Der Abbau in diesem System ergab eine Fragmentierung von Cyclin A mit einem reproduzierbaren Bandenmuster. Diese Ergebnisse sprechen für einen speziellen Abbau durch eine nicht charakterisierte Protease. Zur Untersuchung des Einflusses von Cyclin A auf die Kernzyklen sind RNA Injektionen in 0-1 Stunden alte Drosophila-Embryonen vorgenommen worden, die zur Expression des kompletten, des N-terminal verkürzten oder eines in der destruction box punktmutierten Cyclin A führen sollten. Es konnte gezeigt werden, daß das Cyclin A ∆169 die Kernteilungen inhibiert. Dabei wurden kondensierende Chromosomen beobachtet. Diese sind vermutlich nicht auf einen Block in der Mitose zurückzuführen sondern sind durch eine verfrühte Kondensation der Chromosomen aus der Interphase heraus entstanden. Das Cyclin A ∆169 befindet sich überwiegend im Kern, während ein anderes Konstrukt mit einer Punktmutation (Cyclin AR160G) ausschließlich im Cytoplasma auftritt. Daher kann angenommen werden, daß neben der Stabilität auch die subzelluläre Lokalisation des Cyclin A von Bedeutung ist. 57 6. Literatur Alfa, C.E., Ducommun, B., Beach, D. and Hyams, J.S. (1990). Distinct nuclear and spindle pole body population of cyclin-cdc2 in fission yeast. Nature 347, 680-682. Amon, A., Irniger, S. and Nasmyth, K. (1994). Closing the cell cycle circle in yeast: G2 cyclin proteolysis initiated at mitosis persist until the activation of G1 cyclins in the next cycle. Cell 77, 1037-1050. Booher, R. and Beach, D. (1988). Involvement of cdc13+ in mitotic control in Schizosaccharomyces pombe: Possible interaction of the gene product with microtubules. EMBO J. 6, 3441-3447. Bruce, A.E., Sprenger, F., Duronio, R.J., Leopold, P. and O’Farrell, P.H. (1994). Distinct molecular mechanisms regulate cell cycle timing at successive stages of Drosohila embryogenesis. Gens & Develop. 8, 440-452. De Nooij, J.C., Letendre, M.A. and Hariharan, I.K. (1996). A cyclin-dependent kinase inhibitor, decapo, is necessary for timely exit from the cell cycle during Drosophila embryogeneses. Cell 87, 1237-1247. Debec, A., Kalpin, R.F., Daily, D.R., McCallum, P.D., Rothwell, W.F. and Sullivan W. (1996). Live analysis of free centrosomes in normal and aphicolin-treated Drosophila embryos. J Cell Biol. 134, 103-115. Dong, X., Zavit, K.H., Thomas, B.J., Lin, M., Campbell, S. and Zipursky, S.L. (1997). Control to G1 in the developing Drosophila eye: real regulates Cyclin A. Genes Dev. 11, 94-105. Dower, W., Chassy, B.M., Trevors, J.T. and Blaschek, H.P. (1992). Protocols for the transformation of bacteria by electroporation. In guide to electroporation and electrofusion, edited by Chang, Chassy, Saunders and Sowers. Academic Press Inc., 485-499. Draetta, G. (1997). Cell cycle: will the real Cdk-activating kinase please stand up. Current Biol. 7, R50-R52. Driever, W. and Nüsslein-Vollhard, C. (1988). A gradient of bicoid protein in Drosophila embryos. Cell 54, 83-93. Dunphy, W.G. (1994). The decision to enter mitosis. Trends Cell Biol. 4, 202-207. Duronio, R. and O’Farrell, P.H. (1994). Developmental control af a G1-S transcriptional program in Drosophila. Development 120, 1503-1515. Duronio, R. and O’Farrell, P.H. (1995). Developmental control of the G1 to S transition in Drosophila: cyclin E is a limiting downstream target of E2F. Genes Dev. 9, 1456-1468. 58 Duronio, R., O’Farrell, P.H., Xie, J.E., Brook, A. and Dyson N. (1995). The transcription factor E2F is required for S phase during Drosophila embryogenesis. Genes Dev. 9, 1445-1455. Edgar, B.A. and O’Farrell, P.H. (1989). Genetic control of cell division patterns in the Drosophila embryo. Cell 57, 177-187. Edgar, B.A. and O’Farrell, P.H. (1990). The three postblastoderm cell cycles of Drosophila embryogenesis are regulated in G2 by String. Cell 62, 469-480. Edgar, B.A., Sprenger, F., Duronio, R.J., Leopold, P. and O’Farrell, P.H. (1994). Distinct molecular mechanisms regulate cell cycle timing at successive stages of Drosophila embryogenesis. Genes & Dev. 8, 440-452. Evans, T., Rosenthal, E.T., Youngbloom, J., Distel, D. and Hunt, T. (1983). Cyclin: A protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 33, 389-396. Fisher, R. (1997). CDKs and cyclins in transition(s). Curr. Opin. Gen. & Dev. 7, 32-38. Frohnhöfer, H.G. and Nüsslein-Vollhard, C. (1986). Organisation of anterior pattern in the Drosophila embryo by maternal gene bicoid. Nature 324, 120-125. Frohnhöfer, H.G. and Nüsslein-Vollhard, C. (1987). Maternal genes required for the anterior localization of bicoid activity in the embryo of Drosophila. Genes Dev. 1, 880-890. Foley, E. (1998). Characterization of Roughex - a novel cell cycle inhibitor. Diplamarbeit an der Universität zu Köln. Gabrielli, B.G., Roy, L.M., Gautier, J., Philippe, M. and Maller, J.L. (1992). A Cdc2-related kinase oszillates in the cell cycle indipendendly of cyclins G2/M and Cdc2. J Biol Chem 267, 19691975 Gautier, J., Salomon, M.J., Booher, R.N., Bazan, J.F. and Kirschner, M.W. (1991). Cdc25 is a specific tyrosine phosphatase that directly activates p34cdc2. Cell 67, 197-211. Girard, F., Strausfeld, U., Fernandez, A. and Lamb, N.J. (1991). Cyclin A is required for the onset of DNA replikation in mammalian fibroblasts. Cell 67, 1169-1179. Glotzer, M., Murray, A. and Kirschnerm M.W. (1991). Cyclin is degraded by the ubiquitin pathway. Nature 349, 132-138. Gottlieb, E. (1990). Messenger RNA transport and lokalisation. Curr. Opin. Cell Biol. 2, 10801086. 59 Gould, K.L., Moreno, S., Owen, D.J., Sazer, S. and Nurse, P. (1989). Tyrosine phosphorylation of the fission yeast Cdc2+ protein kinase regulates entry into mitosis. Nature 342, 39-45. Gordon, C., McGurk, G., Dillon, P., Rosen, C. and Hastie, N.D. (1993). Defectiv mitosis due to a mutation in the gene for a fission yeast 26S protease subunit. Nature 366, 355-357. Hagen, I., Hayles, J. and Nurse, P. (1988). Cloning and sequencing of the cyclin related cdc13gene and a cytological study of its role in fission yeast mitosis. J. Cell Sci. 91, 587-595. Hamaguchi, J.R., Tobey, R.A., Pines, J., Crissman, H.A., Hunter, T. and Bradbury, E.M. (1992). Rquirement for p34cdc2 kinase is restricted to mitosis in the mammalian cdc2 mutant FT210. J Cell Biol 117, 1041-1053. Holloway, S.L. Glotzer, M. King, R.W. and Murray, A.W. (1993). Anaphase is initiated by proteolysis rather than by the inactivation of maturation-promoting factor. Cell 73, 1393-402. Hunt, T., Luca, F.C. and Ruderman, J.V. (1992). The requirements for protein synthesis and degradation, and the control of destruction of Cyclin A and B in the meiotic and mitotic cell cycles of the clam embryo. J. Cell Biol. 116, 707-724. Kellogg, D. (1993). The Drosophila mitotic cyclins interact with microtubule-associated proteins. J. Cell Biol. 110, 3244-3254. King R.W., Glotzer, M. and Kirschner, M.W. (1996). Mutagenic analysis of destruction signal of mitotic cyclins and structural characterization of ubiquitinated intermediates. Mol. Biol.of the Cell 7 1343-1357. King, R.W., Deshaies, R.J., Peters, J.M. and Kirschner, M.W. (1996). How proteolysis drives the cell cycle. Science 274, 1652-1659. Knoblich, J.A. and Lehner, C.F. (1993). Synergistic action of Drosophila cyclin A and B during the G2-M transition. EMBO J. 12, 65-74. Knoblich, J.A., Sauer, K., Jones, L., Richardson, H., Saint, R. and Lehner, C.F. (1994). Cyclin E controls S phase progression and is down-regulation during Drosophila embryogenesis is required for arrest of cell proliferation. Cell 77, 107-120. Kobayashi, H., Stewart, E., Poon, R., Adamczewski, J.P., Gannon, J. and Hunt, T. (1992). Identification of the domains in cyclin A required for binding to, and activation of p34cdc2 and p32cdk2 protein kinase subunits. Mol Biol Cell 3, 1279-1294. La Thangue, N.B. (1994). DP and E2F proteins: components of a heterodimeric transcription factor implicated in cell cycle control. Curr Opin Cell Biol 6, 443-450. 60 Larochelle, S., Pandur, J., Fisher, R., Salz, H. and Suter, B. (1998). Cdk7 is essential for mitosis and for in vivo Cdk-aktivating kinase activity. Genes & Dev. 12, 370-381. Lehner, C.F. and O’Farrell, P.H. (1989). Expression and function of Drosophila cyclin A during embryonic cell cycle progression. Cell 56, 957-968. Lehner, C.F. and O’Farrell, P.H. (1990). The Role of Drosophila cyclin A and B in mitotic control. Cell 61, 535-547. Leiss, D., Felix, M.A. and Karsenti, E. (1992). Association of cyclin-bound p34cdc2 with subcellular structures in extracts of Xenopus eggs. J. Cell Sci. 102, 285-297. Lehmann, R. and Nüsslein-Vollhard, C. (1991). The maternal gene nanos has a central role in posterior pattern formation of Drosophila embryo. Development 112, 679-692. Lohka, M., Hayes, M.K. and Maller, J.L. (1988) Purifikation of maturation-promoting factor, an intracellular regulator of early mitotic events. Proc. Natl. Acad. Sci. USA 85, 3009-3013. Lorca, T., Devault, A., Colas, P., Vanloo, A., Fesquet, D., Lazzaro, J.B. and Doree, M. (1992). Cyclin A cys41 does not undergo cell cycle-dependent degradation in Xenopus extracts. FEBS Lett. 306, 90-93. Luca, F.C., Shibuya, E.K., Dohrmann, C.E. and Ruderman, J.V. (1991).Both cyclin A delta 60 and B delta 97 are stable and arrest cells in M-phase, but only cyclin B delta 97 turns on cyclin destruction. EMBO J. 10, 4311-4320. Maldonado-Codina, G. and Glover, D.M. (1992). Cyclins A and B associate with chromatin and the polar regions of spindles, respectively, and do not undergo complete degradation at anaphase in syncycial Drosophila embryos. J. Cell Biol. 116. 967-976. Mac Auley, A., Werb, Z. and Mirkes, P.E. (1993). Characterization of the unusually rapid cell cycles during rat gastrulation. Development 117, 873-883. Macdonald, P.M. and Struhl, G. (1988). Cis-acting sequences responsible for anterior localisation of bicoid mRNA in Drosophila embryos. Nature 336, 595-598. Minshull, J, Blow, J.J. and Hunt, T. (1989). Translation of cyclin mRNA is necessary for activated Xenopus eggs to enter mitosis. Cell 56, 947-956. Minshull, J., Golsteyn, R., Hill, C. and Hunt, T. (1990). The A- and B-type cyclin associated cdc2 kinases in Xenopus turn on and off at different times in the cell cycle. EMBO 8, 2275-2282. Murray, A.W. and Kirschner, M.W. (1989). Cyclin synthesis drives the early embryonic cell cycle. Nature 339, 275-280. 61 Murray, A.W., Solomon, M.J. and Kirschner, M.W. (1989). The role of cyclin synthesis and degradation in the control of maturation promoting factor activity. Nature 339, 280-286. Murray, A. (1995). Cyclin ubiquitination: the destructive end of mitosis. Cell 81, 149-152. Moreno, S., Nurse, P. and Russell, P. (1990). Regulation of mitosis by cyclic accumulation of p80Cdc25 mitotic inducer in fission yeast. Nature 344, 549-552. Morgan, D.O. (1997). Cyclin-dependent kinase: engines, clocks, and microprocessors. Ann Rev Cell Dev Biol 13, 261-291. Nevins, J.R. (1992). E2F: a link between the Rb tumor supressor protein and viral oncoproteins. Science 258, 424-429. Nigg, E.A. (1997). Nucleocytoplamic transport: signals, mechanisms, and regulation. Nature 386, 779-787. Nurse, P. (1990). Universal control mechanism regulating onset of M-Phase. Nature 344, 503-508. Nurse, P. (1994). Ordering S phase and M phase in the cell cycle. Cell 79, 547-550. Ohtsubo, M., Theodoras, A.M., Schumacher, J., Roberts, J.M. and Pagano, M. (1995). Human Cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol Cell Biol 15, 2612-2624. Pagano, M., Pepperkok, R., Verde, F., Ansorge W. and Draetta, G. (1992). Cyclin A is require at two points in the human cell cycle. EMBO J. 11, 961-971. Peter, M. and Herskowitz, I. (1994). Joining the complex: cyclin-dependent kinase inhibitory proteins and the cell cycle. Cell 79, 181-184. Pines, J. and Hunter, T. (1994). The differential localisation of human cyclins A and B is due to a cytoplasmic retention signal in cyclin B. EMBO J. 13, 3772-3781. Raff, J.W. and Glover, D.M. (1988). Nuclear and cytoplasmic mitotic cycles continue in Drosophila embryos in which DNA synthesis is inhibited with Aphidicolin. J. Cell Biol. 107, 20092019. Resnitzky, D., Hengst, L. and Reed, S.I. (1995). Cyclin A-associated kinase activity is rate limiting for entrance into S-phase and is negatively regulated in G1 by p27Kip1. Mol Cell Biol 15, 4347-4352. Richter-Ruoff, B. and Wolf, D.H. (1993). Proteasome and cell cycle. Evidence for a regulatory role of the protease on mitotic cyclins in yeast. FEBS Lett. 336, 34-36. 62 Rosenblatt, J., Gu ,Y. and Morgan, D.O. (1992). Human cyclin-dependent kinase 2 is activated during S and G2 phase of the cell cycle and associates with cyclin A. Proc Natl Acad Sci USA 89, 2824-2848. Russell, P. and Nurse, P. (1986). Cdc25+ functions as an inducer in mitotic control of fission yeast. Cell 45, 145-153. Sauer, K., Knoblich, J.A., Richardson, H. and Lehner, C.F. (1995). Distinct modes of cyclin E/cdc2c kinase regulation and S-phase control in mitotic and endoreduplication cycles of Drosophila embryogenesis. Genes Dev 9, 1327-1339. Sherr, C.J., and Roberts, J.M. (1995). Inhibitors of mammalian G1 cyclin-dependent kinases. Genes & Dev. 7, 1149-1163. Sibon, O.C.M., Stevenson, V.A. and Theurkauf, W.E. (1997). DNA-replication checkpoint control at the Drosophila midblastula transition. Cell 388, 93-97. Siegel, V. and Walter, P. (1988). Each of the activities of signal recognition particle (SRP) is contained within a distinct domain: analysis of biochemical mutans of SRP. Cell 52, 39-49. Sigrist, S., Jacobs, H. Stratmann, R. and Lehner, C.F. (1995). Exit from mitosis is regulated by Drosophila fizzy and the sequential destruction of cyclin A,B and B3. EMBO J. 14, 4827-4838. Schimke, R.T., Kung, A.L., Rush, D.F. and Sherwood, S.W. (1991). Differences in mitotic control among mammalian cells. Cold Spring Harbor Symp. Quant. Biol. 56, 417-425. Sprenger, F., Yakubovich, N., O’Farrell, P.O. (1997). S-phase function of Drosophila cyclin A and its downregultion in G1 phase. Current Biology. 7, 488-499. St. Johnstone, D., Driever, W., Berleth, T., Richstein, S. and Nüsslein-Vollhard, C. (1989). Multible steps in the lokalisation of bicoid RNA to the anterior pole of the Drosophila oocyte. Development 107, 13-19. Stack, J. H. and Newport, J.W. (1997). Developmentally regulated activation of apoptosis early in Xenopus gastrulation results in cyclin A degradation during interphase of the cell cycle. Development 124, 3185-3195 Toyoshima, F., Moriguchi, T., Wada, A., Fukuda, M. and Nishida, E. (1998). Nuclear export of cyclin B and ist possible role in the DNA damage-induced G2 checkpoint. EMBO J. 10, 2728-2735. Tsai, L.H., Harlow, E. and Meyerson, M. (1991). Isolation of the human cdk2 gene that encodes the cyclin A- and adenovirus E1A-associated p33 kinase. Nature 353, 174-177. Van den Heuvel, S. and Harlow, E. (1993). Distinct roles for cyclin-dependent kinases in cell cycle control. Science 262, 2050-2054. 63 Verde, F., Dogterom, M., Stelzer, E., Karsenti ,E. and Leibler, S. (1992). Control of microtuble dynamics and length by Cyclin A- and B-dependent kinases in Xenopus egg extracts. J. Cell Biol. 118, 1097-1108. Walker, D.H. and Maller, J.L. (1991). Role for Cyclin A in the dependence of mitosis on completion of DNA replication. Nature 354, 314-317. Whitfield, W.G.F., Gonzales, C., Maldonado-Codina, G. and Glover, D.M. (1990). The A- and B-type Cyclins of Drosophila are accumulated and destroyed in temporally distinct events that define separable phases of the G2-M transition. EMBO J. 9, 2563-2575.