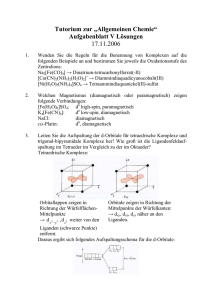
Synthese, Strukturen und Eigenschaften - Ruhr
Werbung

SCHRIFTENREIHE DES MAX-PLANCK-INSTITUTS FÜR STRAHLENCHEMIE ISSN 0932-5131 NR. 139 Übergangsmetallkomplexe neuer ThioetherPhenolatliganden Synthese, Strukturen und Eigenschaften Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften der Fakultät für Chemie an der Ruhr-Universität Bochum vorgelegt von Tobias Kruse Bochum, Januar 2001 Diese Arbeit wurde in der Zeit von März 1998 bis Januar 2001 am MaxPlanck-Institut für Strahlenchemie in Mülheim an der Ruhr angefertigt. Eingereicht am 15.12.2000 Tag der mündlichen Prüfung 30.01.2001 Referent: Prof. Dr. K. Wieghardt Korreferent: Prof. Dr. W. S. Sheldrick Prüfer: Prof. Dr. W. Sander Ich danke allen, die mich durch ihr Interesse und ihre Hilfsbereitschaft unterstützt haben, insbesondere Herrn Prof. Dr. K. Wieghardt für die Stellung des Themas und den mir großzügig gewährten Freiraum bei dessen Bearbeitung Herrn Dr. E. Bill und Dr. K. Hildenbrand für die Anfertigung der EPRSpektren Herrn Dr. Thomas Weyhermüller und Frau Heike Schucht für die Durchführung der Röntgenstrukturanalysen Herrn Jörg Bitter und Frau Kerstin Sand für die Messung unzähliger Kernresonanzspektren Herrn H. Selbach für seinen Einsatz bei den Trennproblemen in der HPLC Frau Dr. Eva Rentschler und Herrn Dr. Frank Nils Penkert für ihre ständige Diskussionsbereitschaft und engagierte Unterstützung bei der Anfertigung dieser Arbeit Herrn Dr. Diran Herebian, Herrn Rüdiger Schumacher, Herrn Udo Beckmann und Herrn Ulrich Schatzschneider für das meiner Arbeit entgegengebrachte Interesse sowie das angenehme Arbeitsklima Inhaltsverzeichnis 1. Einleitung 1 2. Problemstellung 5 3. Die Liganden 6 3.1. Ligandensynthese 6 3.2. Der Ligand H2bse 9 3.3. Der Ligand H2bsb 13 3.4. Der Ligand H2bsbu 17 3.5. Der Ligand Hpsp 20 4. Der Kupferkomplex [Cu2(bse)2] 25 5. Komplexe mit dem Liganden H2bsb 34 5.1. Die Kupferkomplexe [Cu2(bsb)2] und [Cu(bsb)(py)2] 34 5.2. Der Kobaltkomplex [Co(bsb)(Hbsb)] 46 6. Komplexe mit dem Liganden Hpsp 54 6.1. Der Kupferkomplex [Cu(psp)2] 54 6.2. Der Platinkomplex [Pt(psp)2] 59 6.3 Der Palladiumkomplex [Pd(psp)2] 70 6.4. [Pt(psp)2] und [Pd(psp)2] – ein Vergleich 90 7. Komplexe mit dem Liganden H2bsbu 93 7.1. Der Platinkomplex [Pt(bsbu)] 93 7.2. Der Palladiumkomplex [Pd(bsbu)] 98 7.3. Der Kupferkomplex [Cu(bsbu)] 100 8. Zusammenfassung 103 9. Apparativer Teil 107 10. Präparativer Teil 111 11. Anhang 131 12. Literaturverzeichnis 139 Abkürzungen ! Angström = 10-10 m AE Arbeitselektrode B Magnetfeld D Nullfeldaufspaltungsparameter " molarer Extinktionskoeffinzient E Energie, Potential E/D Rhombizität EPR Elektronenspinresonanz Fc/Fc+ Ferrocen/Ferrocenium giso isotroper g-Wert GC Glaskohlenstoff GE Gegenelektrode gg. gegen h Plancksche Konstante HPLC High Pressure Liquid Chromatography I Kernspinquantenzahl IR Infrarot J Kopplungskonstante λ Wellenlänge m/z Masse zu Ladung Verhältnis ν Frequenz NHE Normalenwasserstoffelektrode NMR Kernresonanzspektroskopie ppm Parts per million RE Referenzelektrode S Spin T Temperatur tert Tertiär UV-Vis Ultraviolett/Sichtbar V Volt W EPR-Linienbreite Einleitung 1. 1 Einleitung In vielen Redoxenzymen finden sich in den aktiven Zentren Radikale aromatischer Aminosäuren neben unterschiedlichen Metallionen[1-5]. Zu den redoxaktiven Aminosäureresten gehören vor allem die leicht oxidierbaren Aminosäuren Tryptophan, Cystein und Tyrosin. Ein interessantes Beispiel ist das aktive Zentrum der Galaktose Oxidase (GO), das seit einiger Zeit im Fokus der Forschung steht. Das Enzym wurde erstmals 1957 aus dem Pilz Dactylium dentroides isoliert[6]. Es katalysiert die Oxidation von primären Alkoholen mit Sauerstoff zu den entsprechenden Aldehyden. Hierbei entsteht Wasserstoffperoxid. Die Kristallstruktur wurde 1991 von Ito et al. bestimmt[7]. Tyr495 His496 His581 O HN N N CuII NH O H2O S Tyr272 Cys228 Abb. 1.1: Aktives Zentrum der Galaktose Oxidase In ihrer aktiven Form besitzt das aktive Zentrum der Galaktose Oxidase ein äquatorial an ein Kupfer(II)-Ion koordiniertes Tyrosyl-Radikal. Dieser Tyrosylrest (Tyr272) ist kovalent über eine Thioetherbindung in ortho-Stellung zum phenolischen Sauerstoff mit einem Cystein verknüpft. Über diese Besonderheit ist in der Literatur kontrovers diskutiert worden[8-11]. Arbeiten der letzten Jahre deuten jedoch darauf hin, dass diese Thioethereinheit lediglich eine strukturelle Funktion besitzt. Sie scheint nicht, wie früher vermutet, für eine Erniedrigung des Oxidationspotentials [12, 13] Phenoxylradikals notwendig zu sein . bei der Bildung des Einleitung 2 In den neunziger Jahren wurde eine Vielzahl struktureller Modelle des aktiven Zentrums der Galaktose Oxidase vorgestellt, die Informationen über chemische, strukturelle und physikalische Eigenschaften dieser Spezies erbrachten. Ebenso konnte der Mechanismus des Katalysezyklus aufgeklärt werden[14-22]. Neben strukturellen Modellen sind in den letzten Jahren einige funktionelle Modelle dieses Redoxenzyms publiziert worden, die unter aeroben Bedingungen die Umwandlung primärer Alkohole in die entsprechenden Aldehyde katalysieren. RCH2OH + O2 RCHO + H2O2 Die Abbildung 1.2. zeigt einige dieser katalytisch aktiven Modellsysteme. Alle hier abgebildeten Kupferkomplexe besitzen koordinierte Phenolate, die an ortho- und para-Positionen des Aromaten tert-Butylgruppen tragen. Diese Substituenten tragen zur Stabilisierung der durch Oxidation entstehenden Phenoxyl-Radikale bei. Die tert-Butylgruppe erhöht die Persistenz des Radikals durch sterische Abschirmung. Die Substitution der ortho- und paraPositionen des Phenols mit tert-Butylgruppen ermöglichte die Synthese und Charakterisierung koordinierter Phenoxyl- sowie Aminyl- und ThiylRadikale[23-25]. Sowohl das erste (Abb. 1.2.a) von Stack et al.[26] veröffentlichte Modell, als auch das Modell von Saint-Aman (Abb.1.2.b)[27] und zwei weitere Modelle von Wieghardt et al. (Abb. 1.2.d und 1.2.e)[28, 29] , weisen ein nahezu quadratisch-planar koordiniertes Kupferzentrum mit einem N2O2-Donorsatz auf. Einleitung 3 H H N N CuII O N N O CuII R O O R R´ R´ Abb. 1.2.a Abb. 1.2.b 2+ O S CuII O O CuII O S Abb. 1.2.c + N N O O CuII O Abb. 1.2.d N CuII O NEt3 Abb. 1.2.e Abb. 1.2: Katalysatoren für die aerobe Oxidation von primären Alkoholen Einleitung 4 Nur das ebenfalls von Wieghardt et al. vorgestellte funktionelle Modell 1.2.c besitzt eine dimere Struktur und einen S2O2-Donorsatz[30]. Die Substitution der Thioetherfunktion durch ein Stickstoffatom führt ebenfalls zu einer katalytisch aktiven Spezies (Abb. 1.2.d). Die Effizienz dieser Katalysatoren ist am Beispiel des in Abbildung 1.2.c. gezeigten Modells tabellarisch zusammengefaßt. Substrat Produkt Ausbeute Ethanol Acetaldehyd 63 % Benzylalkohol Benzaldehyd 60 % Tab. 1.1: Ausbeuten der aeroben Oxidation primärer Alkohole Problemstellung 2. 5 Problemstellung Die Besonderheit des von Wieghardt et al. vorgestellten dimeren Kupferkomplexes (Abb. 1.2.c) warf die Frage auf, ob die Substitution der Stickstoffatome durch entsprechende Thioether bei den anderen in Abb. 1.2. aufgeführten funktionellen Modellen ebenfalls zu katalytisch aktiven Spezies führen würde. Es sollte daher die Koordinationschemie anderer Thioether-Phenolatliganden mit Übergangsmetallen systematisch untersucht werden. Der Schwerpunkt dieser Untersuchungen sollte dabei auf den Kupferkomplexen liegen, deren katalytische Aktivität im Sinne eines funktionellen Modells der Galaktose Oxidase überprüft werden sollte. S R OH Abb. 2.1: Schematische Darstellung des neuen Ligandentyps Liganden dieses Typs und deren Metallkomplexe sind nicht bekannt. Nach der Entwicklung eines Syntheseweges für diese Ligandenklasse sind die strukturellen, chemischen und physikalischen Eigenschaften ihrer Übergangsmetallkomplexe von besonderem Interesse. Erstes Ziel war die Darstellung des Schwefelanalogons des ebenfalls von Wieghardt et al. vorgestellten funktionellen Modells (Abb. 1.2.e) der Galaktose Oxidase. Die Liganden 6 3. Die Liganden 3.1. Ligandensynthese Bei der Synthese der neuen Liganden erwies sich die Einführung eines Thioethers in ortho-Position zum Phenolatsauerstoff als besonders schwierig. Diese Position ist für eine Substitution durch eine Thiolat-Einheit wegen des positiven Mesomerie-Effektes des Hydroxy-Substituenten stark desaktiviert. Hier wird die Möglichkeit vorgestellt, durch Oxidation des Schwefels mittels einer Chlorierung und einer gleichzeitigen Metallierung des Phenols zwei sehr reaktive Edukte zur Reaktion zu bringen. Dazu wurde ein allgemeingültiger Syntheseweg für die Darstellung solcher Systeme entwickelt. Folgende Liganden wurden im Rahmen dieser Arbeit synthetisiert: S S S S OH OH HO (H2bse) HO (H2bsb) 1,2-Bis (3,5-di-tert-butyl-2- 1,2-Bis (3,5-di-tert-butyl-2- hydroxy-phenylsulfanyl)-ethan hydroxy-phenylsulfanyl)benzen S S O H (Hpsp) OH S HO (H2bsbu) 4,6-Di-tert-butyl-2- 1,2-Bis (3,5-di-tert-butyl-2- phenylsulfanyl-phenol hydroxy-phenylsulfanyl)-butan Die Liganden 7 Die Synthese dieser vier Liganden gelang nach dem folgenden, allgemeinen Reaktionsschema: Br Br2 OH BuLi RT Abb. 3.1.1: CHCl3 Li OLi -50°C THF OH S Cl-S-R R OLi S H2O RT -60°C Reaktionsschema Br BuLi OH OLi zur Darstellung R der Thioether- Phenolatliganden Die Bromierung des 2,4-Di-tert-butylphenol gelang bei Raumtemperatur in Chloroform in guten Ausbeuten (84%) und hoher Reinheit. Die nachfolgende Metallierung des Aromaten mit Butyllithium-Lösung erfolgte in zwei Schritten. Zunächst wurde bei niedrigen Temperaturen das phenolische Proton substituiert; nach Zugabe eines zweiten Äquivalents Buthyllithiums und zeitgleicher langsamer Erwärmung auf Raumtemperatur wurde das Bromid substituiert. Bei diesem Schritt ist das Arbeiten mit sorgfältig ausgeheizten Glasgeräten und trockenen Lösungsmitteln für eine erfolgreiche Synthese unerlässlich. Da die Buthyllithium-Lösung in exakt stöchiometrischen Mengen zugesetzt wurde, reduzieren insbesondere Spuren von Wasser die Ausbeute dramatisch. Andere Verunreinigungen führten zu unerwünschten, teils hochreaktiven Spezies, die mit den Sulfenylchloriden reagieren. Ein Überschuss an Buthyllithium führte ebenfalls zu einer Reaktion mit den Sulfenylchloriden. Es mußte also bei der Metallierung mit Genauigkeit und Sorgfalt gearbeitet werden. Die Darstellung der Sulfenylchloride erfolgt durch Chlorierung der entsprechenden Thiole mit Sulfurylchlorid[31, 32]. Die Liganden 2 H S 8 SO2Cl2 R R SO2Cl2 S S - HCl Abb. 3.1.2: 2 Cl R S R Reaktionsschema zur Darstellung der Sulfenylchloride Die Umsetzung der unterschiedlichen Thiole und Dithiole mit Sulfurylchlorid erfolgte je nach Stabilität der Sulfenylchloride bei Temperaturen zwischen –30°C und 0°C in trockenem Dichlormethan. Bis zur Hälfte der Zugabe des Sulfurylchlorids wurde eine Gasentwicklung (HCl) beobachtet. Bei den Dithiolen wurde das Entstehen eines polymeren, unlöslichen Feststoffes festgestellt. Dieser löste sich bei weiterer Zugabe von Sulfurylchlorid wieder auf; es entstand das gewünschte Sulfenylchlorid. Besonders die aliphatischen Sulfenylchloride, die keine elektronenziehenden Substituenten tragen, zeichnen sich durch hohe Instabilität aus. Das Benzensulfenylchlorid hingegen konnte durch eine Destillation bei niedrigen Temperaturen (35°C) im Ölpumpenvakuum gereinigt werden. Alle anderen Substrate zersetzen sich bei Reinigungsversuchen und mußten ohne weitere Aufarbeitung eingesetzt werden. Bei der Synthese des Butan-1,4di-sulfenylchlorid wurde ein etwas abgeänderter Weg beschritten[33]. Br Abb. 3.1.3: Br NH4SCN NC S S CN KOH S S Darstellung des 1,2-Dithionats Ausgehend von 1,2-Dithionat erfolgte die Chlorierung mit Sulfurylchlorid in trockenem Diethylether. Die Liganden 3.2. Die 9 Der Ligand H2bse bei der Umsetzung des metallierten Phenols mit Ethan-1,2- disulfenylchlorid bei –50°C in trockenem Diethylether erhaltene Substanz wird aus n-Pentan umkristallisiert. Der Ligand ist in Ausbeuten von über 70% erhältlich. Das Massenspektrum zeigt den M+-Peak bei m/z = 502. Das errechnete Molekulargewicht liegt bei 502.83 g/mol. Das Infrarotspektrum (Abb. 3.2.1.) zeigt bei 3386 cm-1 die typische scharfe Bande für die OH-Valenzschwingung des Phenols. Bei 2965, 2910 und 2870 cm-1 finden sich die CH3-Valenzschwingungen der tert-Butylgruppen. Die aromatischen Ringschwingungen liegen bei 1577 und 1466 cm-1. Die symmetrische Deformationsschwingung der tert-Butylgruppen wird bei 1438 cm-1 und die C-O-Valenzschwingung bei 1175 cm-1 beobachtet. 80 75 70 T [%] 65 60 55 50 45 40 4000 3500 3000 2500 2000 1500 1000 500 -1 W ellenzahl [cm ] Abb. 3.2.1: IR-Spektrum von H2bse Das 1H-NMR- sowie das gemessen. Das 1 13 C-NMR-Spektrum des Liganden wurde in CDCl3 H-NMR-Spektrum ist in Abb. 3.2.2. abgebildet; die Zuordnung der Signale ist in den Tabellen (Tab. 3.2.1. und Tab. 3.2.2.) zusammengefasst. Die Liganden 10 E A S D S OH HO F B ED F B 7,3 7,2 7,1 C 7,0 σ [ppm] 8 7 6 5 A 4 3 2 1 σ [ppm] Abb. 3.2.2: 1 σ [ppm] Multiplizität Integration Zuordnung 1.25 Singulett 18 H tert-Butylgruppe 1.37 Singulett 18 H tert-Butylgruppe 2.78 Singulett 4H CH2-Gruppe 7.03 Singulett 2H OH-Gruppe 7.28 Dublett 2H aromatisches H 7.30 Dublett 2H aromatisches H Tab. 3.2.1: Zuordnung der 1H-NMR-Signale von H2bse; 400 MHz; CDCl3 H-NMR-Spektrum von H2bse; 400 MHz; in CDCl3 0 Die Liganden 11 σ [ppm] Zuordnung σ [ppm] Zuordnung 29.42 CH3; tert-Butylgruppe 117.44 aromatisches C 31.51 CH3; tert-Butylgruppe 126.10 aromatisches C 4 34.29 C ; tert-Butylgruppe 130.30 aromatisches C 35.20 C4; tert-Butylgruppe 135.31 aromatisches C 36.24 CH2-Gruppe 142.35 aromatisches C 153.18 aromatisches C Tab. 3.2.2: Zuordnung der 13C-NMR-Signale von H2bse; 62.9 MHz; CDCl3 Die aus Tetrahydrofuran erhaltenen farblosen Einkristalle von H2bse eigneten sich zur Durchführung einer Röntgenstrukturanalyse. Abb. 3.2.3. zeigt die Struktur des Liganden H2bse und in Tabelle 3.2.3. sind ausgewählte Bindungslängen und –winkel zusammengestellt. C(2) C(15) C(6) Abb. 3.2.3: Kristallstruktur des Liganden H2bse C(3) C(1) C(4) C(5) Die Liganden 12 Bindung Bindungslänge [Å] Winkel S(1)-C(1) 1.781 (3) C(1)-S(1)-C(15) 101.3 (2) S(1)-C(15) 1.847 (4) C(4)-C(3)-C(2) 117.0 (3) O(1)-C(2) 1.380 (4) C(3)-C(4)-C(5) 124.8 (3) C(4)-C(5)-C(6) 116.8 (3) Tab. 3.2.3: Deutlich Bindungswinkel[°] Ausgewählte Bindungslängen und –winkel von H2bse wird aus dieser Struktur die sp3-Hybridisierung des Thioetherschwefelatoms, dessen (C-S-C)-Bindungswinkel mit 101.3° einem leicht gestauchten Tetraederwinkel (109.5°) entsprechen. Weiterhin tritt durch den hohen sterischen Anspruch der tert-Butylgruppen eine Verzerrung der Bindungswinkel im Aromaten auf. An den tert-Butyl substituierten Kohlenstoffatomen (C(3) und C(5)) findet sich ein deutlich gestauchter Winkel (117.0° bzw. 116.8°), wogegen der Winkel am Kohlenstoffatom (C(4)) zwischen diesen beiden gestreckt ist (124.8°). Die Liganden 3.3. 13 Der Ligand H2bsb Die Darstellung des Liganden H2bsb erfolgte durch die Umsetzung des metallierten Phenols mit Benzen-1,2-disulfenylchlorid bei –65°C in trockenem Diethylether. Aufgearbeitet wird die Substanz durch Umkristallisation aus Aceton. Die Ausbeute beträgt 63%. Das Massenspektrum zeigt den M+-Peak bei m/z = 550. Das errechnete Molekulargewicht liegt bei 550.87 g/mol. Das Infrarotspektrum zeigt bei 3408 cm-1 die typische scharfe Bande für die OH-Valenzschwingung des Phenols. Bei 2959, 2910 und 2868 cm-1 finden sich die CH3-Valenzschwingungen der tert-Butylgruppen. Die aromatischen Ringschwingungen liegen bei 1568 und 1469 cm-1. Des weiteren wird die symmetrische Deformationsschwingung der tert-Butylgruppen bei 1441 cm-1 beobachtet; die C-O-Valenzschwingung liegt bei 1182 cm-1. 75 70 T [%] 65 60 55 50 4000 3500 3000 2500 2000 1500 -1 W ellenzahl [cm ] Abb. 3.3.1: IR-Spektrum von H2bsb 1000 500 Die Liganden 14 Das 1H-NMR- sowie das 13 C-NMR-Spektrum des Liganden wurde in CDCl3 1 gemessen. Das H-NMR-Spektrum ist unten abgebildet. D G A S F E G F 7,6 7,4 7,0 HO B C D 7,2 OH E S 6,8 B A 6,6 σ [ppm] 8 7 6 5 4 3 2 1 σ [ppm] Abb. 3.3.2: 1 H-NMR-Spektrum von H2bsb; 500 MHz; in CDCl3 Die Zuordnung der Signale ist in den Tabellen (Tab. 3.3.1. und Tab. 3.3.2.) zusammengefasst. Die nicht zugeordneten Signale werden durch Lösungsmittelmoleküle hervorgerufen. Es gilt folgende Zuordnung: Singuletts bei σ = 1.52 ppm : H2O σ = 2.15 ppm : Aceton σ = 7.24 ppm : CHCl3; 0 Die Liganden 15 σ [ppm] Multiplizität Integration Zuordnung 1.30 Singulett 18 H tert-Butylgruppe 1.40 Singulett 18 H tert-Butylgruppe 6.77 Multiplett 2H aromatische H 6.83 Singulett 2H OH-Gruppe 6.97 Multiplett 2H aromatische H 7.40 Dublett 2H aromatische H 7.43 Dublett 2H aromatische H Tab. 3.3.1: Zuordnung der 1H-NMR-Signale von H2bsb; 500 MHz; in CDCl3 σ [ppm] Zuordnung σ [ppm] Zuordnung 29.42 CH3; tert-Butylgruppe 127.00 aromatisches C 30.92 CH3; tert-Butylgruppe 127.75 aromatisches C 34.42 C4; tert-Butylgruppe 130.91 aromatisches C 35.34 C4; tert-Butylgruppe 135.02 aromatisches C 115.82 aromatisches C 136.03 aromatisches C 126.97 aromatisches C 143.01 aromatisches C 153.45 aromatisches C Tab. 3.3.2: Zuordnung der 13C-NMR-Signale von H2bsb; 125.7 MHz; in CDCl3 Auch bei diesem Liganden gelang es, aus Aceton Einkristalle zu erhalten, die sich für die Durchführung einer Röntgenstrukturanalyse eigneten. Die Struktur ist unten abgebildet (Abb. 3.3.3.). Die Liganden 16 C(18) C(17) C(16) C(19) C(21) C(20) C(6) C(5) C(15) C(1) C(3) C(22) C(4) C(2) Abb. 3.3.3: Kristallstruktur des Liganden H2bsb Bindung Bindungslänge [Å] Winkel Bindungswinkel[°] S(1)-C(1) 1.777 (2) C(1)-S(1)-C(15) 103.25 (10) S(1)-C(15) 1.784 (2) C(20)-S(2)-C(21) 104.06 (10) O(1)-C(2) 1.328 (3) C(4)-C(3)-C(2) 116.6 (2) S(2)-C(21) 1.778 (2) C(3)-C(4)-C(5) 124.9 (2) S(2)-C(20) 1.787 (2) C(4)-C(5)-C(6) 117.0 (2) O(2)-C(22) 1.316 (3) Tab. 3.3.3: Ausgewählte Bindungslängen und –winkel von H2bsb Auch hier zeigen die Bindungswinkel an den beiden Thioetherschwefelatomen wieder deren sp3-Hybridisierung an. Es finden sich Winkel von 103.25 bzw. 104.06°, welches wiederum einem leicht gestauchten Tetraederwinkel (109.5°) entspricht. Die Verzerrung der Bindungswinkel im Aromaten durch die sterisch anspruchsvollen tert-Butylgruppen ist identisch mit der im Liganden H2bse bereits gefundenen. An den tert-Butyl substituierten Kohlenstoffatomen (C(3) und C(5)) findet sich ein deutlich gestauchter Winkel (117.0° bzw. 116.6°), wogegen der Winkel am Kohlenstoffatom (C(4)) zwischen diesen beiden gestreckt ist (124.9°). Die Liganden 3.4. 17 Der Ligand H2bsbu Die in der Einleitung vorgestellten Liganden (Abb. 1.1.d und Abb. 1.1.e) ermöglichen aufgrund ihrer sp2-hybridisierten Stickstoffatome eine planare Koordinationssphäre. Bei den in den Kapiteln 3.2. und 3.3. vorgestellten Liganden H2bse und H2bsb verhindern die sp3-hybridisierten Thioether eine quadratisch-planare Koordinationssphäre. Um mit dieser Geometrie am Thioether einen quadratisch-planar koordinierenden, vierzähnigen Liganden zu realisieren, wird der Ligand H2bsbu mit einer „Brücke“ von vier CH2-Gruppen zwischen den Schwefelatome synthetisiert. Die bei der Umsetzung des metallierten Phenols mit Butan-1,4- disulfenylchlorid bei –80°C in trockenem Diethylether erhaltene Substanz wird mehrfach aus Methanol umkristallisiert. Der Ligand ist in Ausbeuten von 40% erhältlich. Das Massenspektrum zeigt den M+-Peak bei m/z = 530. Das errechnete Molekulargewicht liegt bei 530.88 g/mol. 75 T [%] 70 65 60 55 4000 3500 3000 2500 2000 1500 1000 500 -1 W ellenzahl [cm ] Abb. 3.4.1: Das IR-Spektrum von H2bsbu Infrarotspektrum zeigt die Bande für die symetrische -1 OH- Valenzschwingung des Phenols bei 3379 cm . Bei 2956, 2910 und 2869 cm-1 finden sich die CH3-Valenzschwingungen der tert-Butylgruppen. Die Liganden 18 Die aromatische Ringschwingung liegt bei 1465 cm-1. Die symmetrische Deformationsschwingung der tert-Butylgruppen beobachtet man bei 1438 cm-1. Die C-O-Valenzschwingung liegt bei 1179 cm-1. G D A F S S OH E HO B G F E 7,4 7,3 7,2 7,1 7,0 B 6,9 A σ [ppm] C D 8 7 6 5 4 3 2 1 σ [ppm] Abb. 3.4.2: 1 H-NMR-Spektrum von H2bsbu; 500 MHz; in CDCl3 In CDCl3 beobachtet man neben den Signalen des Liganden H2bsbu noch die Signale des restlichen Methanols. Im 1H-NMR findet sich das Methanolsignal bei σ = 3.47 ppm und im 13C-NMR bei 50.88 ppm. In den Tabellen (Tab. 3.4.1. und Tab. 3.4.2.) ist die Zuordnung der Signale des 1 H-NMR- und des 13C-NMR-Spektrums zusammengefasst. 0 Die Liganden 19 σ [ppm] Multiplizität Integration Zuordnung 1.26 Singulett 18 H tert-Butylgruppe 1.37 Singulett 18 H tert-Butylgruppe 1.66 Multiplett 4H CH2-Gruppe 2.63 Multiplett 4H CH2-Gruppe 7.06 Singulett 2H OH-Gruppe 7.27 Dublett 2H aromatische H 7.31 Dublett 2H aromatische H Tab. 3.4.1: Zuordnung der 1H-NMR-Signale von H2bsbu; 500 MHz; in CDCl3 σ [ppm] Zuordnung σ [ppm] Zuordnung 28.48 CH2-Gruppe 118.61 aromatisches C 29.44 CH3; tert-Butylgruppe 125.73 aromatisches C 31.53 CH3; tert-Butylgruppe 130.22 aromatisches C 34.29 C4; tert-Butylgruppe 135.05 aromatisches C 35.19 C4; tert-Butylgruppe 142.11 aromatisches C 36.40 CH2-Gruppe 152.98 aromatisches C Tab. 3.4.2: Zuordnung der 13C-NMR-Signale von H2bsbu; 125.8 MHz; CDCl3 Die Liganden 3.5. 20 Der Ligand Hpsp Mit dem Ziel, durch zweizähnige Liganden eine quadratisch-planare Koordinationssphäre zu ermöglichen, wurde der Ligand Hpsp synthetisiert. Das Benzensulfenylchlorid ist relativ stabil, und konnte sogar bei niedrigen Temperaturen destillativ aufgearbeitet werden. Erhalten wurde der Ligand Hpsp aus der Umsetzung des zuvor metallierten Phenols mit dem Benzensulfenylchlorid in trockenem Diethylether bei –65°C. Das Produkt wird durch Destillation gereinigt und kristallisiert langsam bei Raumtemperatur aus dem erhaltenen hochviskosen Öl aus. Die Ausbeute liegt bei ca. 65 %. Das Massenspektrum zeigt den M+-Peak bei m/z = 314. Das errechnete Molekulargewicht liegt bei 314.49 g/mol. Das Infrarotspektrum zeigt bei 3391 cm-1 die typische scharfe Bande für die OH-Valenzschwingung des Phenols. Bei 2962, 2910 und 2867 cm-1 finden sich die CH3-Valenzschwingungen der tert-Butylgruppen. Die aromatischen Ringschwingungen liegen bei 1581 und 1478 cm-1. Des weiteren beobachtet man die symmetrische Deformationsschwingung der tert-Butylgruppen bei 1440 cm-1. Die C-O-Valenzschwingung liegt bei 1181 cm-1. 70 65 T [%] 60 55 50 45 4000 3500 3000 2500 2000 1500 -1 W ellenzahl [cm ] Abb. 3.5.1: IR-Spektrum von Hpsp 1000 500 Die Liganden 1 Die 21 13 H- und C-NMR-Spektren wurden in deuteriertem Chloroform aufgenommen. Das 1H-NMR-Spektrum ist abgebildet (Abb. 3.5.2.); alle Zuordnungen sind in den Tabellen (Tab. 3.5.1. und Tab. 3.5.2.) zusammengefasst. E H A S D G O H C B G H 7,6 7,4 F D E 7,2 7,0 F C 6,8 σ [ppm] 8 7 Abb. 3.5.2: B 6 1 5 4 σ [ppm] 3 2 A 1 H-NMR-Spektrum von Hpsp; 400 MHz; in CDCl3 0 Die Liganden 22 σ [ppm] Multiplizität Integration Zuordnung 1.29 Singulett 9H tert-Butylgruppe 1.41 Singulett 9H tert-Butylgruppe 6.84 Singulett 2H OH-Gruppe 7.04 Dublett 2H aromatische H 7.13 Triplett 1H aromatisches H 7.22 Triplett 2H aromatische H 7.31 Dublett 2H aromatische H 7.41 Dublett 2H aromatische H Tab. 3.5.1: Zuordnung der 1H-NMR-Signale von Hpsp; 400 MHz; in CDCl3 σ [ppm] Zuordnung σ [ppm] Zuordnung 29.41 CH3; tert-Butylgruppe 126.82 aromatisches C 31.49 CH3; tert-Butylgruppe 129.07 aromatisches C 34.36 C4; tert-Butylgruppe 131.03 aromatisches C 35.27 C4; tert-Butylgruppe 135.73 aromatisches C 115.57 aromatisches C 136.43 aromatisches C 125.71 aromatisches C 142.77 aromatisches C 126.30 aromatisches C 153.39 aromatisches C Tab. 3.5.2: Zuordnung der 13C-NMR-Signale von Hpsp; 62.9 MHz; CDCl3 Die Liganden 23 Die aus Methanol erhaltenen farblosen Einkristalle eigneten sich zur Durchführung einer Röntgenstrukturanalyse. Die Struktur ist unten abgebildet (Abb. 3.5.3.). C(19) C(18) C(20) C(15) C(6) C(5) C(17) C(16) C(1) C(4) C(3) C(2) Abb. 3.5.3: Kristallstruktur des Liganden Hpsp In Tabelle 3.5.3. sind ausgewählte geometrische Daten der Struktur von Hpsp aufgelistet. Bindung Bindungslänge [Å] Winkel S(1)-C(1) 1.774 (3) C(1)-S(1)-C(15) 104.63 (13) S(1)-C(15) 1.775 (3) C(3)-C(4)-C(5) 124.3 (3) O(1)-C(2) 1.385 (4) C(4)-C(3)-C(2) 117.0 (2) C(4)-C(5)-C(6) 116.6 (2) Tab. 3.5.3: Bindungswinkel[°] Ausgewählte Bindungslängen und –winkel von Hpsp Die Liganden 24 Der Ligand Hpsp zeigt die gleichen Strukturmerkmale wie die zuvor beschriebenen Liganden H2bse und H2bsb. Auch hier findet man wieder den typischen sp3-hybridisierten Thioether mit einem Bindungswinkel von 104.63° zu den beiden Kohlenstoffatomen sowie die Verzerrung der Bindungswinkel im Aromaten durch die sterisch anspruchsvollen tert-Butylgruppen. Der Kupferkomplex [Cu2(bse)2] 4. 25 Der Kupferkomplex [Cu2(bse)2] Die Darstellung des Kupferkomplexes des Liganden H2bse erfolgte durch die Umsetzung des Liganden mit Cu(I)Cl in Tetrahydrofuran mit Triethylamin als Base und anschließender Oxidation an Luft. Im Massenspektrum wurde der Molekülpeak bei m/z = 1128 beobachtet (berechnet: 1128.71 g/mol). Dies entspricht nicht der monomeren Form [Cu(bse)], sondern der einer dimeren Spezies [Cu2(bse)2]. Die durch Kristallisation aus Aceton erhaltenen Einkristalle, die sich für die Durchführung einer Röntgenstrukturanalyse eigneten, bestätigen die Ergebnisse der Massenspektroskopie. Es handelt sich um ein Dimer, bei dem jeweils einer der Phenolatsauerstoffe verbrückend koordiniert ist. Die Struktur ist unten abgebildet (Abb. 4.1.). Abb. 4.1: Kristallstruktur des Komplexes [Cu2(bse)2] Der Kupferkomplex [Cu2(bse)2] 26 Bindung Bindungslänge [Å] Winkel Cu(1) – Cu(1A) 2.8140 (10) S(1)-Cu(1)-O(1) 78.74 (8) Cu(1) – S(2) 2.3733 (12) S(1)-Cu(1)-O(1A) 130.05 (8) Cu(1) – S(1) 2.6221 (12) O(1A)-Cu(1)-S(2) 139.79 (8) Cu(1) – O(1) 1.927 (3) O(1)-Cu(1)-O(2) 168.78 (12) Cu(1) – O(1A) 2.023 (3) S(2)-Cu(1)-O(2) 88.00 (9) Cu(1) – O(2) 1.861 (3) Cu(1)-O(1)-Cu(1A) 90.82 (10) Tab. 4.1: Bindungswinkel[°] Ausgewählte Bindungslängen und –winkel in [Cu2(bse)2] Die Kupferzentren befinden sich in einer verzerrt quadratisch-pyramidalen Koordinationsumgebung. Die Schwefelatome S(1) und S(1A) besetzen die apikalen Positionen. Auffällig bei der Struktur dieses Kupferkomplexes ist der kurze Abstand zwischen den beiden Kupferzentren (2.814 Å) sowie der ungewöhnlich kleine Winkel Cu-O-Cu am überbrückenden Phenolatsauerstoff (90.8°). Außerdem weist der überbrückende Phenolatsauerstoff O(1) keine planare sp2-Hybridisierung auf, sondern eine deutliche Verzerrung in Richtung sp3-Hybridisierung. Abb. 4.2: Geometrie am überbrückenden Sauerstoff des Komplexes [Cu2(bse)2] Der Kupferkomplex [Cu2(bse)2] 27 Definiert man drei Ebenen durch a) beide Kupferzentren und den überbrückenden Sauerstoff (Cu(1)-O(1)Cu(1A)) b) den Sauerstoff, den an ihn gebundenen Kohlenstoff C(2) sowie Cu(1) und c) den Sauerstoff, den an ihn gebundenen Kohlenstoff C(2) sowie Cu(1A). sollten die Winkel α und α´ zwischen den Ebenen (a)(b) und (a)(c) bei einem sp2-hybridisierten Sauerstoff gleich null, die Ebenen somit koplanar sein. Die in diesem Fall gefundenen Werte zeigen eine deutliche Abweichung von der Koplanarität. Es ist: α = 45.3° und α´ = 50.1°. Diese für phenoxo-, aber auch alkoxo- und hydroxo-überbrückte Kupferdimere ungewöhnlichen strukturellen Eigenschaften haben starken Einfluss auf die magnetische Kopplung der Spins der beiden Kupferzentren miteinander. Die Messung der temperaturabhängigen magnetischen Suszeptibilität zeigt eine ferromagnetische Kopplung der beiden Kupferzentren. (Abb. 4.3.) Die Temperaturabhängigkeit von µeff konnte mit folgenden Parametern simuliert werden: H = - 2J S1 S2 S1 = S2 = ½ g1 = g2 = 1.99 J12 = +36.5 cm-1 TIP = 142 10-6 cm-3 mol-1 Der Kupferkomplex [Cu2(bse)2] 28 2,85 2,80 µ eff / µ b 2,75 2,70 2,65 2,60 0 50 100 150 200 250 300 Tem peratur / K Abb. 4.3: Magnetmessung des Komplexes [Cu2(bse)2] bei 1 T Die Wechselwirkung zwischen zwei Kupferatomen, die jeweils einen Spinzustand S =1/2 besitzen, führt zu zwei molekularen Zuständen. Zum einen zu einem Spin-Singulett (S = 0) und zum anderen zu einem Spin-Triplett (S=1). Die Energiedifferenz zwischen diesen beiden Zuständen ist 2J. Für den Fall, dass die Wechselwirkung antiferromagnetisch ist, ist der Grundzustand S=0 und J negativ; wenn die Wechselwirkung ferromagnetisch ist, folgt ein Grundzustand S = 1 und J ist positiv. Der Singulett-Triplett-Abstand kann vereinfacht verstanden werden, als die Summe aus antiferromagnetischer, negativer ferromagnetischer, positiver Komponente JF[34]. J = JAF + JF Komponente JAF und Der Kupferkomplex [Cu2(bse)2] 29 Für den Fall, dass die beiden Kupferzentren identisch sind, wird die antiferromagnetische Komponente JAF durch das Überlappungsintegral zwischen den beiden magnetischen Orbitalen (dx²-y²) ferromagnetische Komponente JF der Kopplung bestimmt, die hingegen durch das Zweielektronen-Austauschintegral. Der Betrag der antiferromagnetischen Komponente ist hierbei meist deutlich größer als der Betrag des ferromagnetischen Anteils. Für die in diesem Fall beobachtete ferromagnetischen Kopplung muß also das Überlappungsintegral sehr klein sein, so dass der Anteil JF überwiegt und man den oben beschriebenen Triplett-Grundzustand beobachten kann. In der Literatur finden sich verschiedene Arbeiten, die sich mit der Korrelation zwischen Struktur und magnetischer Eigenschaft von überbrückten Kupferdimeren beschäftigen. So veröffentlichte Hatfield 1976 erstmals eine systematische Untersuchung an hydroxo-überbrückten Kupferdimeren, bei denen eine lineare Korrelation zwischen dem Abstand der beiden Kupferzentren sowie des Brückenwinkels am Sauerstoff und den magnetischen Eigenschaften der Komponenten erkannt wurde[35]. Bei Verringerung des Abstandes und Verkleinerung des Brückenwinkels wurde der antiferromagnetische Anteil der Kopplung kleiner. Bei einer Variation des Winkels Cu-O-Cu von 95.6 auf 104.1° verändert sich die Kopplung zwischen den Kupferzentren von +172 nach –509 cm-1[35]. Auch für alkoxo- und andere sauerstoffüberbrückte Kupferdimere findet sich ein vergleichbarer Effekt[36]. Der Austausch findet vorwiegend über die Brückenatome unter Beteiligung der dx²-y²-Orbitale der Metallzentren und der sund p-Orbitale des Sauerstoffs statt[37]. Eine out-of-plane-Verzerrung an den Kupferzentren in Richtung des apikal gebundenen Liganden führt somit zu einer Verringerung des σ–Overlaps und damit zu einer Verringerung des Austausches. Dieser Effekt ist jedoch nur sehr klein[38]. Weiterhin besteht eine Korrelation zwischen dem Grad der pyramidalen Verzerrung Brückensauerstoffs Komponente[39-41]. und der des normalerweise Verkleinerung der sp2-hybridisierten antiferromagnetischen Der Kupferkomplex [Cu2(bse)2] 30 Diese Beobachtung wird von Bertrand[42] und Gatteschi[43] durch einen zweiten Austauschweg zwischen den beiden Metallzentren erklärt. Demnach existiert ein π–Overlap zwischen den out-of-plane-Kupfer-Orbitalen mit dem nicht bindenden pz-Orbital des Brückensauerstoffs. Bei einer Verzerrung der trigonalen Planarität am Sauerstoff wird dieser Austausch verringert. Es folgt eine Abnahme der antiferromagnetischen Komponente mit zunehmender Verzerrung der sp2-Geometrie am Brückensauerstoff. Zusammenfassend kann man diese verschiedenen Einflüsse der Struktur auf die magnetischen Eigenschaften von sauerstoffüberbrückten Kupferdimeren wie folgt beschreiben: Die antiferromagnetische Komponente der Wechselwirkung verringert sich 1) mit abnehmendem Kupfer-Kupfer Abstand. 2) mit abnehmendem Cu-O-Cu-Brückenwinkel 3) mit dem Grad der pyramidalen Verzerrung am Brückensauerstoff 4) mit zunehmendem Abstand des Kupferzentrums von der Koordinationsebene Da der unter 4) aufgeführte Effekt jedoch sehr klein ist, beschränke ich mich in diesem Fall auf die Betrachtung der ersten drei aufgeführten Einflüsse. Man findet in im Falle des [Cu2(bse)2] einen kurzen Cu-Cu-Abstand (2.814 Å), einen sehr kleinen Brückenwinkel (90.8°), sowie eine deutliche Verzerrung der trigonalen Planarität am überbrückenden Phenolatsauerstoff (α = 45.3 bzw. 50.1°). Das Zusammenspiel dieser drei strukturellen Eigenschaften des hier untersuchten Kupferkomplexes verringert den antiferromagnetischen Anteil der Kopplung drastisch und es ergibt sich in der Summe die hier zu beobachtende ferromagnetische Kopplung zwischen den beiden Metallzentren (J = +36.5 cm-1). Der Kupferkomplex [Cu2(bse)2] Die Elektrochemie des 31 Kupferkomplexes [Cu2(bse)2] in entgastem Dichlormethan zeigt zwei irreversible Oxidationen. -0,5 -1,0 I [µ A] -1,5 -2,0 -2,5 -3,0 1200 1000 800 600 400 U [mV] vs Fc/Fc Abb. 4.4: 200 0 + Square-wave-Voltammogramm des Komplexes [Cu2(bse)2] in CH2Cl2 (0.1 M TBAPF6); T = 298 K; Frequenz = 30 Hz Die Potentiale liegen bei +520 und +710 mV gegen Fc/Fc+. Ob es sich hierbei um ligandenzentrierte oder metallzentrierte Oxidationen handelt, kann aufgrund der Instabilität der oxidierten Komplexe nicht geklärt werden. Diese sind einer weitergehenden, spektroskopischen Untersuchung nicht zugänglich. Der Kupferkomplex [Cu2(bse)2] 32 Das UV-Vis-Spektrum des Kupferkomplexes [Cu2(bse)2] zeigt eine für ein äquatorial an ein quadratisch-pyramidales Kupferzentrum koordiniertes Phenolat typische Ligand-Metall-charge-transfer-(LMCT)-Bande bei 450 nm (ε =3300 l mol-1 cm-1). Zusätzlich beobachtet man eine Bande bei 645 nm (ε = 1450 l mol-1 cm-1), die einem d-d-Übergang zugeordnet wird[44]. 4 2 3 -1 -1 ε [10 l*mol *cm ] 3 1 0 400 500 600 700 800 900 1000 1100 λ [nm ] Abb. 4.5: UV-Vis-Spektren des Komplexes [Cu2(bse)2] in CH2Cl2 vor (durchgezogene Linie) und nach Zugabe von zwei Äquivalenten Pyridin (gepunktete Linie) Bei der Zugabe von Pyridin als koordinierende Base verändert sich das Spektrum wie in der Literatur für vergleichbare dimere Kupferkomplexe beschrieben[45-47]. Man erkennt eine deutliche Abnahme der Intensität beider Banden und deren gleichzeitige bathochrome Verschiebung. In diesem Fall verschiebt sich die charge-transfer-Bande um 14 nm auf 464 nm (ε = 1200 l mol-1 cm-1) und die d-d-Bande um über 100 nm auf 769 nm (ε = 680 l mol-1 cm-1). Der Kupferkomplex [Cu2(bse)2] 33 Eine Isolierung des monomeren Komplexes, bei dem das Pyridin koordiniert ist, gelingt nicht. Aus den verschiedenen Ansätzen kristallisiert der dimere Komplex [Cu2(bse)2]. Dass sich in diesem Fall jedoch eine monomere Spezies bildet, zeigt der Vergleich dieser UV-Vis-Spektren mit den Spektren des im nächsten Kapitel (Kap. 5.1.1.) vorgestellten Komplexes [Cu2(bsb)2]. Die Spektren zeigen in allen Bereichen vor und nach der Zugabe von Pyridin Übereinstimmung. Es findet sich die gleiche bathochrome Verschiebung der charge-transfer- und der d-d-Bande bei gleichzeitiger Intensitätsabnahme der beiden Banden. Bei dem Komplex [Cu2(bsb)2] gelingt im Gegensatz zu dem hier besprochenen Kupferkomplex jedoch die Isolierung des monomeren Komplexes. Die erhaltenen Einkristalle eignen sich zur Durchführung Röntgenstrukturanalyse und beweisen die monomere Struktur. einer Komplexe mit dem Liganden H2bsb 5. Komplexe mit dem Liganden H2bsb 5.1. Die Kupferkomplexe [Cu2(bsb)2] und [Cu(bsb)(py)2] 34 Der Kupferkomplex [Cu2(bsb)2] wurde durch Umsetzung des Liganden H2bsb mit Cu(I)Cl und Triethylamin als Base in Tetrahydrofuran und anschließender Oxidation an Luft, dargestellt. Im Massenspektrum findet man den Molekülpeak bei m/z = 1224. Dies entspricht analog zum Kupferkomplex [Cu2(bse)2] einer dimeren Spezies [Cu2(bsb)2]. Die durch Kristallisation aus Dichlormethan erhaltenen Einkristalle, die sich für die Durchführung einer Röntgenstrukturanalyse eigneten, bestätigen auch in diesem Fall die Ergebnisse der Massenspektroskopie. Es handelt sich wiederum um ein Dimer, bei dem jeweils einer der Phenolatsauerstoffe verbrückend koordiniert ist. Die Struktur ist abgebildet (Abb. 5.1.1.). Auch hier befinden sich die Kupferzentren in einer verzerrt quadratischpyramidalen Koordinationsumgebung. Die Schwefelatome S(2) und S(2A) besetzen die apikalen Positionen. Ausgewählte Bindungslängen und –winkel sind in der Tabelle 5.1.1. zusammengefasst. Komplexe mit dem Liganden H2bsb Abb. 5.1.1: 35 Kristallstruktur des Komplexes [Cu2(bsb)2] Bindung Bindungslänge [Å] Winkel Cu(1) – Cu(1A) 2.904 (2) S(1)-Cu(1)-O(1) 88.1 (2) Cu(1) – S(2) 2.766 (2) S(1)-Cu(1)-O(2A) 166.9 (2) Cu(1) – S(1) 2.308 (2) O(1)-Cu(1)-S(2) 134.5 (2) Cu(1) – O(1) 1.877 (5) O(1)-Cu(1)-O(2) 151.3 (2) Cu(1) – O(1A) 1.961 (5) S(2)-Cu(1)-O(2) 74.3 (2) Cu(1) – O(2) 1.943 (5) Cu(1)-O(2)-Cu(1A) 96.1 (2) Tab. 5.1.1: Bindungswinkel[°] Ausgewählte Bindungslängen und –winkel in [Cu2(bsb)2] Komplexe mit dem Liganden H2bsb Abb. 5.1.2: 36 Geometrie am überbrückenden Sauerstoff des Komplexes [Cu2(bsb)2] Der Kupfer-Kupfer-Abstand (2.904 Å) ist auch in diesem Fall kurz. Der Bindungswinkel am Brückensauerstoff (96.1°) ist ebenfalls klein. Eine Untersuchung der pyramidalen Verzerrung am überbrückenden Phenolatsauerstoff lässt auch in diesem Fall eine deutliche Abweichung von der trigonal-planaren Geometrie erkennen (Abb. 5.1.2.). Die Winkel α und α´ betragen in diesem Fall 47.1° und 49.1° und weichen somit um etwa den gleichen Betrag von der Koplanarität ab, wie in Kapitel 4 für den Komplex [Cu2(bse)2] beschrieben. In Analogie lassen die in Kapitel 4 besprochenen Korrelationen zwischen strukturellen und magnetischen Eigenschaften eine ferromagnetische Kopplung für den Komplex [Cu2(bsb)2] erwarten. Komplexe mit dem Liganden H2bsb 37 Die Messung der temperaturabhängigen magnetischen Suszeptibilität von [Cu2(bsb)2] bestätigt diese Erwartung. 3.00 2.95 µ eff / µ B 2.90 2.85 2.80 2.75 2.70 0 50 100 150 200 250 300 Tem peratur / K Abb. 5.1.3: Magnetmessung des Komplexes [Cu2(bsb)2] bei 1 T Die Temperaturabhängigkeit von µeff konnte mit folgenden Parametern simuliert werden: H = -2J S1 S2 S1 = S2 = 1/2 g1 = g2 = 2.10 J12 = + 58.7 cm-1 Komplexe mit dem Liganden H2bsb 38 Die elektrochemischen Untersuchungen des Kupferkomplexes [Cu2(bsb)2] in entgastem Dichlormethan zeigen zwei irreversible Oxidationen. -0,5 -1,0 I [µ A] -1,5 -2,0 -2,5 -3,0 -3,5 0 200 400 600 800 U [m V] vs Fc/Fc Abb. 5.1.4: 1000 1200 1400 + Square-wave-Voltammogramm des Komplexes [Cu2(bsb)2] in CH2Cl2 (0.1 M TBAPF6); T = 298 K; Frequenz = 30 Hz Die Potentiale liegen bei +620 und +990 mV gegen Fc/Fc+. Ob es sich hierbei um ligandenzentrierte oder metallzentrierte Oxidationen handelt, kann aufgrund der Instabilität der oxidierten Komplexe nicht geklärt werden. Diese waren einer zugänglich. weitergehenden, spektroskopischen Untersuchung nicht Komplexe mit dem Liganden H2bsb 39 Das UV-Vis-Spektrum des dimeren Komplexes [Cu2(bsb)2] weist zwei charakteristische Banden auf. Zum einen die Phenolat-Kupfer-charge-transfer(LMCT)-Bande bei 470 nm (ε = 2320 l mol-1 cm-1) und zum anderen die einem d-d-Übergang zugeordnete Bande bei 620 nm (ε = 630 l mol-1 cm-1). 3,0 2,5 -1 ε [10 l*m ol *cm ] 2,0 -1 1,5 3 1,0 0,5 0,0 -0,5 400 500 600 700 800 900 1000 1100 λ [nm ] Abb. 5.1.5: UV-Vis-Spektren des Komplexes [Cu2(bsb)2] in CH2Cl2 vor (durchgezogene Linie) und nach Zugabe von zwei Äquivalenten Pyridin (gepunktete Linie) Bei Zugabe von zwei Äquivalenten Pyridin pro dimerem Komplex verändert sich das Spektrum in gleicher Weise, wie es bei dem in Kapitel 4 vorgestellten, dimeren Kupferkomplexen [Cu2(bse)2] beobachtet wird. Es erfolgt eine deutliche Farbänderung der Lösung von braun nach hellviolett. Auch hier erkennt man eine Abnahme der Intensität beider Banden, sowie deren gleichzeitige bathochrome Verschiebung. Die charge-transfer-Bande verschiebt sich hier auf 517 nm (ε = 580 l mol-1 cm-1). Die d-d-Bande befindet sich nach Zugabe des Pyridins bei 743 nm (ε = 250 l mol-1 cm-1). Komplexe mit dem Liganden H2bsb 40 Die Daten deuten auf die Entstehung einer monomeren Spezies [Cu(bsb)(py)], in der sich das Kupferzentrum in einer quadratisch-pyramidalen Koordinationssphäre befindet, hin. Die Darstellung eines monomeren Kupferkomplexes mit koordiniertem Liganden H2bsb erfolgte durch Lösen des dimeren Komplexes [Cu2(bsb)2] in Dichlormethan und Zugabe eines Überschusses Pyridin. Das UV-Spektrum ändert sich dabei nach der Zugabe des zweiten Äquivalents nicht mehr. Der auskristallisierte Feststoff zeigt im Massenspektrum zunächst die Freisetzung von Pyridin und dann einen Molekülpeak bei m/z = 611 ([Cu(bsb)]+). Der Komplex ist also unter den Bedingungen der Massenspektroskopie nicht stabil. Die zunächst beobachtete Freisetzung des Pyridins spricht jedoch für das Vorhandensein des PyridinAdukts. Auch Fukuzumi[46] berichtet über die Instabilität der Pyridin-Adukte ähnlicher dimerer Ausgangsverbindungen unter den Bedingungen der Massenspektroskopie. Das EPR-Spektrum des monomeren Komplexes zeigt in Übereinstimmung mit der Literatur[48-50] ein axiales S = ½ Signal bei giso = 2.13. Deutlich sichtbar ist die Hyperfein-Kopplung zum Kernspin 3 /2 des Kupfers. Sim. Exp. dX´´ dB 100 200 300 400 500 B [mT] Abb. 5.1.8: Experimentelles und simuliertes EPR-Spektrum des Komplexes [Cu(bsb)(py)2] in CH2Cl2 T = 10 K; Frequenz: 9.4368 GHz; Leistung: 10.1 µW; Modulation: 1.25 mT Komplexe mit dem Liganden H2bsb 41 Simuliert werden kann das in Abb. 5.1.8. gezeigte EPR-Spektrum des monomeren Komplexes mit folgendem Parametersatz: gx = 2.06 Wx = 19 mT gy = 2.11 Wy = 23 mT gz = 2.22 Wz = 10 mT Inuc = 1.5 Az = 15 mT Zum Vergleich ist das EPR-Spektrum des dimeren Ausgangsproduktes abgebildet (Abb. 5.1.9.), das eine deutlich stärkere Anisotropie aufweist. Es handelt sich um ein rhombisches S = 1-Spektrum mit einem giso-Wert von 2.09. Dieses verdeutlicht die starke Verzerrung der quadratisch-pyramidalen Koordinationssphäre der Kupferionen im dimeren Komplex [Cu2(bsb)2]. Sim. dX´´ dB Exp 250 300 350 400 B [mT] Abb. 5.1.9: Experimentelles und simuliertes EPR-Spektrum des Komplexes [Cu2(bsb)2] in CH2Cl2 T = 10 K; Frequenz: 9.4365 GHz; Leistung:100.8 µW; Modulation: 0.99 mT Komplexe mit dem Liganden H2bsb 42 Für die Simulation des EPR-Spektrums des dimeren Kupferkomplexes [Cu2(bsb)2] wurden folgende Parameter verwendet: gx = 2.15 Wx = 6.0 mT Ax = 12 mT gy = 2.107 Wy = 7.0 mT Ay = 0 mT gz = 2.02 Wz = 4.0 mT Az = 2.5 mT S=1 Inuc = 1.5 -1 D = -0.006 cm E = 0.002 cm-1 Aus Dichlormethan kristallisiert der Komplex [Cu(bsb)(py)2] in hellvioletten Einkristallen aus, die sich zur Durchführung einer Röntgenstrukturanalyse eigneten Die Struktur ist in Abb. 5.1.6. gezeigt. Abb. 5.1.6: Kristallstruktur des Komplexes [Cu(bsb)(py)2] Komplexe mit dem Liganden H2bsb 43 Ausgewählte Abstände und Bindungswinkel sind in der Tabelle 5.1.2. zusammengefaßt. Bindung Bindungslänge [Å] Winkel Cu(1) – O(1) 1.929 (3) S(1)-Cu(1)-S(2) 81.65 (5) Cu(1) – O(2) 1.932 (3) O(1)-Cu(1)-O(2) 178.97 (13) Cu(1) – S(1) 2.422 (2) N(1)-Cu(1)-N(2) 101.09 (14) Cu(1) – S(2) 2.696 (2) S(2)-Cu(1)-N(1) 164.13 (10) Cu(1) – N(1) 2.394 (4) N(1)-Cu(1)-O(1) 91.34 (14) Cu(1) – N(2) 2.067 (4) O(1)-Cu(1)-N(2) 89.5 (2) Tab. 5.1.2: Bindungswinkel[°] Ausgewählte Bindungslängen und –winkel in [Cu(bsb)(py)2] Überraschend bei der Struktur des isolierten monomeren Kupferkomplexes [Cu(bsb)(py)2] ist die Tatsache, das hier zwei Pyridine an ein Kupferzentrum koordiniert sind und sich somit eine für ein Kupfer(II)-Zentrum recht ungewöhnliche oktaedrische Koordinationssphäre ergibt. Wie für ein oktaedrisch koordiniertes Cu(II)-Ion (d9) erwartet, erkennt man eine deutliche Jahn-Teller-Verzerrung.[51-53] Es handelt sich in diesem Fall um einen tetragonal verzerrten, gestreckten Oktaeder. Die Bindungen zu den axial koordinierten Stickstoff- und Schwefelatomen (N(1) und S(2)) sind signifikant länger als die Abstände zu den gleichartigen Donoratomen N(2) und S(1), die äquatorial koordiniert sind. Das hier beobachtete EPR-Spektrum des sechsfach koordinierten Kupferzentrums ist nahezu identisch mit den Spektren der fünffach koordinierten Komplexe, die in der Literatur beschrieben werden. Für eine Jahn-Teller-verzerrte, oktaedrische Koordinationssphäre am Kupfer wird wie für ein quadratisch-pyramidal koordiniertes Kupferion ein axiales Spektrum erwartet. Komplexe mit dem Liganden H2bsb 44 Ebenso erkennt man im Elektronenanregungsspektrum nach Zugabe zweier Äquivalente Pyridin pro dimerem Komplex [Cu2(bsb)2] keine weitere Änderung des Spektrums bei weiterer Zugabe von Pyridin mehr. Um die monomere Spezies [Cu(bsb)(py)2] zu erhalten muß jedoch ein deutlicher Überschuss Pyridin zugegeben werden. Durch die Jahn-Teller-Verzerrung am oktaedrisch koordinierten Kupferzentrum kann also weder durch die EPR- noch durch die UV-VisSpektroskopie der fünffach koordinierte, quadratisch-pyramidale Komplex [Cu(bsb)(py)] von dem sechsfach koordinierten, oktaedrischen Komplex [Cu(bsb)(py)2] unterschieden werden. Es entsteht in diesem Fall zunächst bei der Zugabe zweier Äquivalente Pyridin ein quadratisch-pyramidal koordinierter Kupferkomplex. Bei weiterer Zugabe von Pyridin wandelt sich dieser in einen sechsfach koordinierten, oktaedrischen Komplex [Cu(bsb)(py)2] um, der wie oben beschrieben in kristalliner Form erhalten wird. Die Isolierung der fünffach koordinierten Spezies [Cu(bsb)(py)] gelang nicht. Komplexe mit dem Liganden H2bsb 45 S O Cu S O O Cu O S S + 2 Py S 2 O S + 2 Py Cu S 2 N N Cu S O O Abb. 5.1.7: O N Umwandlung des dimeren Komplexes [Cu2(bsb)2] in die monomeren Komplexe [Cu(bsb)(py)2] und[Cu(bsb)(py)2] durch Zugabe von Pyridin in Dichlormethan Komplexe mit dem Liganden H2bsb 5.2. 46 Der Kobaltkomplex [Co(bsb)(Hbsb)] Durch die Umsetzung von einem Äquivalent des Liganden H2bsb mit einem Äquivalent Co(II)Cl2 und zwei Äquivalenten Triethylamin in trockenem THF wurde, nach Oxidation an Luft, der braune Kobaltkomplex [Co(bsb)(Hbsb)] erhalten. Durch Umkristallisation aus Diethylether/Chloroform wurden Einkristalle erhalten, die sich zur Durchführung einer Röntgenstrukturanalyse eigneten. Die Struktur ist unten in Abb. 5.2.1. gezeigt. Abb. 5.2.1: Kristallstruktur des Komplexes [Co(bsb)(Hbsb)] Komplexe mit dem Liganden H2bsb 47 Die folgende Tabelle fasst ausgewählte Abstände und Bindungswinkel zusammen. Bindung Bindungslänge [Å] Winkel Co(1) – O(2) 1.872 (4) S(2)-Co(1)-S(4) 168.57 (6) Co(1) – O(3) 1.892 (4) O(3)-Co(1)-S(3) 87.87 (14) Co(1) – O(4) 1.898 (4) O(2)-Co(1)-O(4) 90.4 (2) Co(1) – S(2) 2.239 (2) S(4)-Co(1)-S(3) 92.44 (10) Co(1) – S(3) 2.217 (2) S(4)-Co(1)-O(2) 86.11 (13) Co(1) – S(4) 2.211 (2) S(4)-Cu(1)-O(3) 92.29 (13) Tab. 5.1.2: Bindungswinkel[°] Ausgewählte Bindungslängen und –winkel in [Co(bsb)(Hbsb)] Das Kobalt(III)ion befindet sich in einer oktaedrischen Koordinationssphäre. Die axialen Positionen werden von den beiden Schwefelatomen S(2) und S(4) eingenommen. Es koordinieren zwei Liganden an ein Kobaltzentrum, wobei einer der Liganden nur zweizähnig mit einem Phenolatsauerstoff und einer Thioether-Funktion koordiniert ist. Die anderen beiden Donoratome sind nicht koordiniert. Das nicht koordinierte Phenol ist protoniert. Das Kobalt(III)Zentrum (d6 low spin) ist facial von drei Sauerstoff- und drei Schwefelatomen umgeben. Alle Sauerstoff-Kobalt-Bindungen sind kürzer als 1.90 Å. Diese kurzen Bindungsabstände finden sich auch in anderen Phenolat-Kobalt(III)-Spezies[54, 55] . Die Bindungsabstände der Thioether zum Kobaltion sind kurz. Es finden sich jedoch in der Literatur einige Beispiele mit ähnlich kurzen ThioetherKobalt(III)-Bindungsabständen[56-60]. Sowohl die Messung der temperaturabhängigen magnetischen Suszeptibilität als auch das 1H-NMR-Spektrum belegen den diamagnetischen Grundzustand des Komplexes [Co(bsb)(Hbsb)]. Komplexe mit dem Liganden H2bsb 48 Das Cyclovoltammogramm des Kobaltkomplexes [Co(bsb)(Hbsb)] in entgastem Dichlormethan (Abb. 5.2.2.) zeigt eine reversible, ligandenzentrierte Oxidation bei +450 mV gegen Fc/Fc+. Dieses vergleichsweise niedrige Potential erklärt sich durch die vollständige Besetzung der t2g-Orbitale des d6 low-spin Systems beim oktaedrisch koordinierten Kobalt(III). Es findet keine π–Rückbindung vom Sauerstoff des Phenolats zum Metallzentrum statt. Die Elektronendichte ist somit am Sauerstoff sehr hoch, so dass eine Oxidation zum Phenoxyl-Radikal bei relativ niedrigem Potential erfolgt. 4 2 0 I [ µ A] -2 -4 -6 -8 -10 -12 700 600 500 400 300 E [m V] gg. Fc/Fc Abb. 5.2.2: 200 100 0 + Cyclovoltammogramm des Komplexes [Co(bsb)(Hbsb)] bei 50, 100, 200, 400 mV/s; in CH2Cl2 (0.1 M TBAPF6); T = 298 K Die oxidierte Spezies erweist sich auf der Zeitskala der Coulometrie bei –25°C und kleinen Konzentrationen als stabil, so dass sie einer spektroskopischen Untersuchung zugänglich war. Komplexe mit dem Liganden H2bsb 49 Die potentialkontrollierte Coulometrie des Komplexes [Co(bsb)(Hbsb)] erfolgt in entgastem Dichlormethan bei –25°C. Als Leitsalz wird Tetrabutylammonium-hexafluorophosphat mit einer Konzentration von 0.1 mol/l verwendet. Die Konzentration des Kobaltkomplexes beträgt 0.2 x 10-4 mol/l. Der Verlauf der elektrochemischen Oxidation wird mit Hilfe der UV-VisSpektroskopie verfolgt. Abbildung 5.2.3. zeigt die während der Oxidation aufgenommen Elektronenanregungsspektren. 35000 25000 20000 ε [l mol-1 cm-1] 30000 25000 15000 10000 0 20000 320 340 360 380 400 λ [nm] 420 440 5000 15000 4000 ε [l mol-1 cm-1] ε [l mol-1 cm-1] 5000 10000 3000 2000 1000 5000 0 500 0 200 300 400 500 600 700 600 800 700 800 λ [nm] 900 900 1000 1100 1000 λ [nm] Abb. 5.2.3: UV-Vis-Spektren während der Coulometrie des Komplexes [Co(bsb)(Hbsb)] in CH2Cl2 (0.1 M TBAPF6); T = 248 K; c = 2 x 10-4 mol/l 1100 Komplexe mit dem Liganden H2bsb 50 Das UV-Vis-Spektrum des nicht oxidierten Komplexes [Co(bsb)(Hbsb)] zeigt im UV-Bereich zwei Absorptionen bei 245 (ε = 3.1 104 l mol-1 cm-1) und 282 (ε = 2.75 104 l mol-1 cm-1) nm und eine Schulter bei 314 nm (ε = 2.2 104 l mol-1 cm-1). Diese Absorptionen werden π-π*-Übergängen des aromatischen Systems zugeordnet. Im sichtbaren Bereich befinden sich Absorptionsmaxima bei 407 nm (ε = 1.0 103 l mol-1 cm-1) und 778 nm (ε = 0.49 103 l mol-1 cm-1). Diese entsprechen zwei d-d-Übergängen. Für low-spin Kobalt(III) im oktaedrischen Feld mit einem 1A1g-Grundtherm werden zwei spin erlaubte d-d-Übergänge erwartet[61]. Die energieärmere Bande bei 778 nm wird dem 1 T1g←1A1g-Übergang zugeordnet, die energiereichere Bande bei 407 nm dem 1T2g←1A1g-Übergang. Während der elektrochemischen Oxidation erscheinen drei neue Banden bei 400 nm (ε = 7.6 103 l mol-1 cm-1), bei 510 nm (ε = 2.7 103 l mol-1 cm-1) und bei 830 nm (ε = 1.1 103 l mol-1 cm-1). Außerdem erkennt man die leichte Abnahme der Bande bei 314 nm sowie einen isosbestischen Punkt bei 351 nm. Es findet sich die typische Phenoxyl-Radikalbande bei 400 nm. Diese ist gegenüber den in der Literatur beschriebenen um ca. 15 nm hypsochrom verschoben[23, 54]. Die beiden anderen Banden können hingegen nicht dem Phenoxyl-Radikal zugeordnet werden. Es finden sich aber deutliche Übereinstimmungen zu den typischen Banden von koordinierten Schwefelradikalen. Kimura[25] berichtet bei einem, ebenfalls an ein Kobalt(III)-Zentrum koordinierten, Thiylradikal von zwei Banden bei 509 (ε = 2.6 103 l mol-1 cm-1) und 784 (ε = 1.0 103 l mol-1 cm-1) nm. Diese Übereinstimmung sowohl in der Lage der Banden, als auch in ihren Intensitäten, lässt den Schluss zu, dass ein wesentlicher Teil des Radikals auf dem Schwefelatom des Thioethers lokalisiert ist. Komplexe mit dem Liganden H2bsb 51 R R S R´ R´ S Co R´ S Co O Co O R´ R´ Abb. 5.2.4. R O R´ Mesomere Grenzstrukturen der oxidierten Spezies [Co(bsb)(Hbsb)]+• Es wird also eine Mischung aus koordiniertem Schwefel- und koordiniertem Phenoxyl-Radikal beobachtet. Die Aufenthaltswahrscheinlichkeit des ungepaarten Elektrons bei thioether-substituierten Phenoxyl-Radikalen ist sowohl experimentell, als auch durch theoretische Rechnungen bestimmt worden[62, 63]. Demnach ist das ungepaarte Elektron zu einem großen Teil in den pz-Orbital des Phenolat-Sauerstoffs (0.19) und des Schwefel (0.29), sowie zu kleineren Teilen an den aromatischen Kohlenstoffen in der ortho- und para-Position zum Sauerstoff (0.08-0.17) lokalisiert. Das Elektronenanregungsspektrum der oxidierten Spezies [Co(bsb)(Hbsb)]+• besitzt große Übereinstimmung mit den Spektren der Radikalform von 2Methyl-thio-p-cresol[λmax = 400 und 830 nm)[64, 65] und der oxidierten Form der Galaktose Oxidase (λmax = 445 und 800 nm)[66]. Das X-Band EPR-Spektrum des oxidierten Komplexes [Co(bsb)(Hbsb)]+•, das in einer 0.3 mm Flachzelle bei Raumtemperatur gemessen wurde, zeigt ein S = 1 /2-Signal bei g = 2.0204 (Abb. 5.2.5.). Dieses steht im Einklang mit der Formulierung eines organischen Radikals und zeigt die erwartete Hyperfein-Kopplung dieses organischen Radikals zum Kobaltkern mit dem Kernspin 7/2. Komplexe mit dem Liganden H2bsb 52 Das isotrope Spektrum kann mit folgenden Parametern simuliert werden: Inuc = 7/2 giso = 2.0204 Wiso = 0.251 mT ACo = 0.78 mT Die Kopplung des Radikals zum Kobaltkern ist mit 0.78 mT etwas größer, als sie für reine Phenoxyl-Radikale gefunden wird (0.5 – 0.6 mT)[55, 67] , jedoch deutlich kleiner als sie für koordinierte Semichinone beschrieben wird (0.9 – 1.2 mT)[68-71]. Für das an ein Kobalt(III)-Zentrum koordinierte Thiyl-Radikal findet Kimura[25] eine Kopplung von 1.1 mT. Abb. 5.2.5: Experimentelles und simuliertes EPR-Spektrum des Komplexes [Co(bsb)(Hbsb)]+• in CH2Cl2 T = 298 K; Frequenz: 9.4528 GHz; Leistung: 5 mW; Modulation: 0.85 mT Diese Hyperfein-Kopplung zum Kobaltkern beweist, dass das koordinierte Phenolatanion des vierfach koordinierten Liganden in [Co(bsb)(Hbsb)] zum Phenoxyl-Radikal oxidiert wird und nicht etwa der unkoordinierte Phenolteil des koordinierten (Hbsb)-- Liganden. Komplexe mit dem Liganden H2bsb 53 Bei der elektrochemischen Oxidation des Komplexes [Co(bsb)(Hbsb)] erhält man einen Radikal-Komplex [Co(bsb)(Hbsb)]+•. Das Radikal ist zu einem Großteil auf den beiden Donoratomen (Phenolatsauerstoff und Thioether) lokalisiert und nur zu geringen Teilen über den aromatischen Ring delokalisiert. Das Elektronenanregungsspektrum der oxidierten Spezies entspricht in großen Teilen des Spektrums der oxidierten Form der Galaktose Oxidase, obwohl in diesem Fall kein Kupfer, sondern ein Kobaltion das Metallzentrum bildet. Komplexe mit dem Liganden Hpsp 6. Komplexe mit dem Liganden Hpsp 6.1. Der Kupferkomplex [Cu(psp)2] 54 Der aus der Umsetzung von zwei Äquivalenten des Liganden Hpsp und einem Äquivalent Cu(I)Cl unter Zusatz von zwei Äquivalenten Triethylamin in Methanol synthetisierte Komplex [Cu(psp)2] wurde aus der Reaktionslösung in Form brauner Einkristalle erhalten, die sich zur Durchführung einer Röntgenstrukturanalyse eigneten. Die Struktur ist in Abb. 6.1.1. abgebildet. Abb. 6.1.1: Kristallstruktur des Komplexes [Cu(psp)2] Komplexe mit dem Liganden Hpsp 55 Bindung Bindungslänge [Å] Winkel Bindungswinkel[°] Cu – S(1) 2.3511 (9) S(1)-Cu-O(1) 87.53 (7) Cu – S(2) 2.3395 (9) S(1)-Cu-S(2) 161.05 (3) Cu – O(1) 1.846 (2) O(1)-Cu-O(2) 165.37 (10) Cu – O(2) 1.851 (2) S(1)-Cu-O(2) 94.78 (7) Tab. 6.1.1: Ausgewählte Bindungslängen und –winkel in [Cu(psp)2] Das Kupfer(II)ion befindet sich in einer verzerrten, quadratisch-planaren Koordinationssphäre. Bezogen auf die Orientierung der beiden an das Metallzentrum bindenden Schwefelatome liegt der Komplex in der transKonfiguration vor. Die fünfte Koordinationsstelle oberhalb der Ebene wird durch einen Thioether des Nachbarkomplexes besetzt. Der Abstand zum Schwefelatom in der apikalen Position beträgt 3.13 Å. Der Cu-Cu-Abstand beträgt 4.37 Å. Es ergibt sich eine dimere Struktur. Aufgrund der großen Entfernung der beiden paramagnetischen Zentren voneinander erwartet man nur eine sehr kleine ferromagnetische Komponente der Kopplung zwischen den beiden Kupferzentren. Die Orthogonalität der magnetischen dx²-y²-Orbitale an den Kupferionen zu dem sp3-Orbital, des apikalen Schwefels, verhindert eine antiferromagnetische Wechselwirkung zwischen den beiden Zentren. Die Messung der temperaturabhängigen magnetischen Suszeptibilität zeigt die erwartete schwache ferromagnetische Kopplung zwischen den beiden Kupferionen (Abb. 6.1.2.). Komplexe mit dem Liganden Hpsp 56 2,7 µ eff / µ B 2,6 2,5 2,4 0 50 100 150 200 250 300 Tem perature / K Abb. 6.1.2: Magnetmessung des Komplexes [Cu(psp)2] bei 1 T Die Temperaturabhängigkeit von µeff kann mit folgenden Parametern simuliert werden: S1 = S2 = 1/2 g1 = g2 = 2.07 J12 = + 0.5 cm-1 TIP = 51 x 10-6 cm-3 mol-1 (pro Dimer) Komplexe mit dem Liganden Hpsp 57 Das UV-Vis-Spektrum der Verbindung [Cu(psp)2], aufgenommen in CH2Cl2, zeigt fünf Absorptionsmaxima bei 245 (ε = 4.0 104 l mol-1 cm-1), 290 (ε = 1.7 104 l mol-1 cm-1), 320 (ε = 1.25 104 l mol-1 cm-1), 360 (ε = 0.5 104 l mol-1 cm-1) und 440 (ε = 0.3 104 l mol-1 cm-1) nm. 4 2 4 ε [10 l m ol -1 -1 cm ] 3 1 0 200 400 600 800 1000 λ [nm ] Abb. 6.1.3: UV-Vis-Spektrum des Komplexes [Cu(psp)2] in CH2Cl2 Die Banden unter 400 nm werden den π-π*-Übergängen des Aromaten zugeordnet, die Bande bei 440 nm ist ein für ein äquatorial an ein Kupferzentrum koordiniertes Phenolat typischer Ligand-zu-Metall-ChargeTransfer-(LMCT)-Übergang. Komplexe mit dem Liganden Hpsp Die Elektrochemie des 58 Kupferkomplexes [Cu(psp)2] in entgastem Dichlormethan zeigt vier irreversible, ligandenzentrierte Oxidationen bei +500, +570, +800 und +950 mV gegen Fc/Fc+. 0 -1 I [µ A] -2 -3 -4 -5 -6 1200 1000 800 600 E [mV] gg. Fc/Fc Abb. 6.1.4: 400 200 0 + Square-wave-Voltammogramm des Komplexes [Cu(psp)2] in CH2Cl2; Frequenz: 30 Hz Jeweils zwei der beobachteten Oxidationen liegen bei ähnlichem Potential. Das Auftreten von vier anstelle der erwarteten zwei ligandenzentrierten Oxidationen, lässt darauf schließen, dass in Lösung zwei in ihrem Oxidationspotentialen sehr ähnliche Komponenten vorliegen. Ein Vergleich mit dem elektrochemischen Verhalten des Palladiumkomplexes [Pd(psp)2] dieses Liganden läßt den Schluß zu, das es sich hierbei um cis-trans-Isomere des Kupferkomplexes handelt (vgl. Seite 89). Die Oxidationen bei +500 und +800 mV gegen Fc/Fc+ werden dem cis-Isomer zugeordnet. Die Potentialdifferenz entspricht mit ∆E = 300 mV dem ∆E, welches für die cisIsomere des Palladium- und Platinkomplexes des gleichen Liganden gefunden wurde. Die Potentialdifferenz der Oxidationen des trans-Isomers (+ 570 mV und + 950 mV gegen Fc/Fc+) ist mit ∆E = 380 mV dem bei dem trans[Pd(psp)2] gefundenen vergleichbar. Komplexe mit dem Liganden Hpsp 6.2. 59 Der Platinkomplex [Pt(psp)2] Der aus der Umsetzung von PtCl2 mit zwei Äquivalenten des Liganden Hpsp unter dem Zusatz von NaOH erhaltene Komplex [Pt(psp)2] kristallisiert aus der Reaktionslösung in gelben Einkristallen aus, die zur Durchführung einer Röntgenstrukturanalyse geeignet waren. Die abgebildete Kristallstruktur (Abb. 6.2.1.) zeigt das Platin(II)ion in einer quadratisch-planaren Koordinationssphäre. Ausgewählte Bindungslängen und –winkel sind in der Tabelle 6.2.1. zusammengefasst. Abb. 6.2.1: Kristallstruktur des Komplexes [Pt(psp)2] Komplexe mit dem Liganden Hpsp 60 Bindung Bindungslänge [Å] Winkel Bindungswinkel[°] Pt – S(1) 2.2589 (14) S(1)-Pt-O(1) 85.86 (13) Pt – S(2) 2.2595 (14) S(1)-Pt-S(2) 104.12 (5) Pt – O(1) 1.990 (4) O(1)-Pt-O(2) 84.1 (2) Pt – O(2) 1.986 (4) S(1)-Pt-O(2) 169.97 (12) Tab. 6.2.1: Ausgewählte Bindungslängen und –winkel in [Pt(psp)2] Die beiden Liganden sind cis-ständig bezogen auf die Orientierung der beiden Schwefeldonoren an das Metallzentrum gebunden. Die ebenfalls denkbare trans-Konfiguration wird bei diesem Komplex weder in Lösung noch im Kristall beobachtet. Auf diese Bevorzugung der cis-Konfiguration bei der Koordination „weicher“ Liganden (wie z. B. Thioethern) an ein Metallion der Klasse b (z. B. PtII, PdII), wird im Kapitel 6.4. detaillierter eingegangen. Die Gegenwart von zwei pyramidal koordinierten Thioethern führt zu zwei möglichen diastereomeren Formen. Am Schwefel findet sich entweder R- oder S-Konfiguration. Bei der in Abb. 6.2.1. gezeigten Struktur wird für das Schwefelatom S(1) R-Konfiguration gefunden, für das Schwefelatom S(2) SKonfiguration. Die beiden Phenylsubstituenten am Schwefel befinden sich auf der gleichen Seite der Koordinationsebene. Im weiteren wird diese Konfiguration als (syn) bezeichnet. Für den Fall, das an beiden Schwefelatomen die gleiche Konfiguration vorliegt, befinden sich die Phenylsubstituenten auf gegenüberliegenden Seiten selbiger Ebene (anti). Diese beiden enantiomeren Formen ((R,R) und (S,S)) sind im NMRExperiment nicht unterscheidbar. Während im Kristall ausschließlich die syn-Konfiguration zu beobachten ist, lassen sich in Lösung beide Diastereomere im NMR-Experiment nachweisen. Für jede tert-Butylgruppe der Liganden finden sich im 1 H-NMR bei Raumtemperatur zwei Signale. Es findet also in Lösung eine Inversion am Schwefel statt. Es handelt sich hierbei um eine intramolekulare, reversible Umwandlung zwischen der syn- und der anti-Form mit einer Kinetik erster Ordnung. Komplexe mit dem Liganden Hpsp 61 k anti syn k´ Für die Geschwindigkeitskonstante k gilt die Eyring-Gleichung RT − k= e NA ⋅ h ∆G # RT (Gleichung 6.2.1.) ∆G# freie Aktivierungsenthalpie R Gaskonstante 8.31441 J K-1 mol-1 NA Avogadrosche Konstante 6.02205 1023 mol-1 h Plancksche Konstante 6.62618 10-34 J s Es gilt: Bei einer auf der Zeitskala eines NMR-Experiments (10-1 bis 10-9s) langsamen Umwandlungen beobachtet man die Signale von beiden Spezies. Eine schnelle Umwandlung liefert hingegen nur ein mittleres Signal. Zwischen dem Bereich des schnellen und des langsamen Austausches treten breite Absorptionen auf[72]. Durch Variation der Temperatur kann so die Koaleszenstemperatur (Tc) bestimmt werden, bei der die getrennten Signale zusammenfallen. An diesem Punkt gilt für die Geschwindigkeitskonstante kTc: k Tc = π υ A −υB 2 υ A −υB (Gleichung 6.2.2.) Frequenzdifferenz der beiden Signale bei tiefen Temperaturen Mittels der Methode der Koaleszenztemperaturbestimmung kann also die Inversionsbarriere der Umwandlung am Schwefel bestimmt werden. Komplexe mit dem Liganden Hpsp 62 Durch Einsetzen von Gleichung 6.2.2. in Gleichung 6.2.1. und anschließender Umformung erhält man folgende Beziehung: ∆G ≠ = R ⋅ Tc ⋅ ln R ⋅ Tc 2 π ⋅ N A ⋅ hυ A − υ B (Gleichung 6.2.3.) Misst man Tc in K und die Absorption ν in Hz, dann erhält man: ∆G≠ = 19.1 10-3 Tc(9.97 + logTc – logνA - νB) (Gleichung 6.2.4.) Bei dem Komplex [Pt(psp)2] kann die Koaleszenstemperatur für beide tertButylgruppensignale ermittelt werden. Hierzu wird die Substanz in deuteriertem Dimethylformamid gelöst und es werden 1H-NMR-Spektren bei unterschiedlichen Temperaturen aufgenommen. Die Intensitätsverhältnisse der Signale blieben über den gesamten Temperaturbereich nahezu gleich bei etwa 4:5. Es liegt also in Lösung keine deutliche Dominanz der syn- oder anti-Konfiguration vor. Die auch im Kristall beobachtete syn-Konfiguration ist wegen der π-Stapelung der Phenylringe vermutlich die thermodynamisch bevorzugte Form. Jede der beiden tert-Butylgruppen zeigt im 1H-NMR-Spektrum wie oben erwähnt bei Raumtemperatur zwei Signale. Bei einer Erhöhung der Temperatur ist das Zusammenfallen der beiden, jeweils zu einer tert-Butylgruppe gehörenden, Signale deutlich zu erkennen. Da die Frequenzdifferenzen (νA - νB) der Signale der beiden tertButylgruppen bei Raumtemperatur nicht gleich sind, ergeben sich aus obiger Gleichung (Gl. 6.2.4.) auch zwei verschiedene Koaleszenztemperaturen Tc. Abbildung 6.2.2. zeigt den Ausschnitt der 1H-NMR-Spektren mit den Signalen der tert-Butylgruppen bei unterschiedlichen Temperaturen. Die Koaleszenztemperatur wird für die ortho zum Phenolatsauerstoff stehende tert-Butylgruppe bei 343 K und für die para-ständige tert-Butylgruppe bei 348 K bestimmt. Komplexe mit dem Liganden Hpsp Abb. 6.2.2: 1 63 H-NMR des Komplexes [Pt(psp)2] in Dimethylformamid-d7 Bei Frequenzdifferenzen von 5.5 Hz für die ortho-ständige tert-Butylgruppe (σ1 = 1.53 ppm) und 6.5 Hz für die para-ständige (σ2 = 1.19 ppm) ergibt sich eine freie Aktivierungsenthalpie von 77.2 kJ/mol bzw. 77.9 kJ/mol. Im Rahmen der Messgenauigkeit kann die freie Aktivierungsenthalpie für die Inversion am Schwefel angegeben werden zu: ∆G# = 77.5 +/- 1 kJ/mol Für vergleichbare Platinkomplexe finden sich in der Literatur Werte von 56 bis 89 kJ/mol[73-76]. Die freie Aktivierungsenthalpie der Inversion am Schwefel ist von verschiedenen Faktoren abhängig. Der sterische Anspruch der Substituenten, das Metallzentrum, die Elektronendichte am Metall sowie die am Inversionszentrum. Bei zyklischen Thioethern spielt auch die Ringgröße eine entscheidenede Rolle. Diese Einflüsse werden in Kap. 6.4. diskutiert. Komplexe mit dem Liganden Hpsp 64 Das Cyclovoltammogramm des Komplexes [Pt(psp)2] zeigt in Dichlormethan zwei reversible ligandenzentrierte Oxidationen bei +670 und +990 mV gegen Fc/Fc+. Abb. 6.2.3: Cyclovoltammogramm des Komplexes [Pt(psp)2] bei 20, 50, 100, 200, 400 mV/s; in CH2Cl2 (0.1 M TBAPF6); T = 298 K Die Potentialdifferenz der beiden Oxidationen liegt mit ∆E = 320 mV in dem Bereich, der in der Literatur für zwei aufeinanderfolgende ligandenzentrierte Oxidationen bei Phenolaten gefunden wird[54, 77]. Diese Potentialdifferenz kann durch ein einfaches elektrostatisches Modell erklärt werden. Hierbei werden lediglich die Solvatationsenergien der unterschiedlich geladenen Teilchen berücksichtigt. Laut Born-Gleichung[6.2.7.] (Gl. 6.2.5.) ist die Energie sphärischer Partikel mit dem Radius r von seiner Ladung n und der Dielektrizitätskonstante εoεE des Lösungsmittels abhängig[79]. Komplexe mit dem Liganden Hpsp n 2e2 N L − ∆G 0 = E f ⋅ F = 8πrε 0 ε E Ef Redoxpotential in Volt F Faraday-Konstante NL Avogadrozahl 65 (Gleichung 6.2.5.) Die Potentialdifferenz zweier aufeinanderfolgender Oxidationen kann somit berechnet werden zu: e2 N L ∆E =( 2n − 1) ⋅ 8πrε 0 ε E F f (Gleichung 6.2.5.) Die Auftragung der Redoxpotentiale gegen die Ladung des Liganden liefert eine Gerade, deren Steigung für ein gegebenes Substitutionsmuster des Liganden typisch ist und nur wenig von der Art des Metallions abhängt. Da das Platin unter diesen Bedingungen nicht oxidiert wird, wird wie bei dem Kobaltkomplex [Co(bsb)(Hbsb)] bei der ersten Oxidation der entsprechende Radikal-Komplex [Pt(psp)2]+• erzeugt. Ob bei der zweiten Oxidation die diradikalische Spezies [Pt(psp)2]2+•• oder eine chinoide Spezies entsteht, kann hier nicht entschieden werden, da die zweifach oxidierte Spezies auf der Zeitskala der Coulometrie nicht stabil ist (vgl. Abb. 6.2.4.). Eine Untersuchung dieser Substanz ist demnach nicht möglich. Komplexe mit dem Liganden Hpsp R 66 R R S S - e- S Pt O R S Pt + e- O O O - eR R + e- S S Pt O Abb. 6.2.4: O mögliche Oxidationsprodukte des Komplexes [Pt(psp)2] Das Monoradikal-Kation erweist sich auf der Zeitskala der Coulometrie bei tiefen Temperaturen und geringen Konzentrationen als stabil, so das es einer weiteren spektroskopischen Untersuchung zugänglich ist. Die Coulometrie des Komplexes [Pt(psp)2] erfolgt in entgastem Dichlormethan bei –20°C. Als Leitsalz wird Tetra-n-butylammoniumhexafluorophosphat mit einer Konzentration von 0.1 mol/l verwendet. Die Konzentration des Platinkomplexes beträgt 0.2 x 10-4 mol/l. Der Verlauf der potentialkontrollierten Coulometrie wird mit Elektronenanregungsspektroskopie verfolgt (Abb. 6.2.5.). Hilfe der Komplexe mit dem Liganden Hpsp 67 30 30 3 ε [103 l*mol-1*cm-1] 25 ε [103 l*mol-1*cm-1] 20 ε [103 l*mol-1*cm-1] 25 20 15 10 5 0 250 260 270 280 290 300 310 320 330 λ [nm] 15 2 1 0 400 500 600 700 800 λ [nm] 900 1000 1100 10 5 0 200 300 400 500 600 700 800 900 1000 1100 λ [nm] Abb. 6.2.5: UV-Vis-Spektren während der Coulometrie des Komplexes [Pt(ptb)2] in CH2Cl2 (0.1 M TBAPF6); T = 253 K; c = 2 x 10-4 mol/l Das UV-Vis-Spektrum des nicht oxidierten Komplexes zeigt im sichtbaren Bereich keine Absorptionsmaxima. Im UV-Bereich befinden sich zwei Absorptionen bei 250 (ε = 3.0 104 l mol-1 cm-1) und 330 (ε = 7.0 103 l mol-1 cm-1) nm, sowie zwei Schultern bei 270 (ε = 2.3 104 l mol-1 cm-1) und 380 (ε = 0.5 103 l mol-1 cm-1) nm. Letztere ist für die schwach gelbe Färbung des Komplexes verantwortlich. Die Banden unter 350 nm werden π-π*-Übergängen des aromatischen Systems zugeordnet, die intensitätsschwache Absorption bei 380 nm hingegen einem d-d-Übergang. Komplexe mit dem Liganden Hpsp 68 Während der elektrochemischen Oxidation beobachtet man eine Abnahme der Absorption bei 270 nm und zeitgleich das Auftreten mehrerer Absorptionsbanden im UV- wie auch im sichtbaren Bereich des Spektrums. Es finden sich neue Absorptionen bei 300 (ε = 1.2 104 l mol-1 cm-1), 380 (ε = 1.9 103 l mol-1 cm-1), 460 (ε = 1.8 103 l mol-1 cm-1), 570 (ε = 1.9 103 l mol-1 cm-1) und 690 (ε = 2.25 103 l mol-1 cm-1) nm. Bei 278 nm findet sich ein isosbestischer Punkt. Die Banden bei 300 und 460 nm werden einem Schwefelradikal zugeordnet. Tripathi[80] und Asmus[81] berichten beide über Adsorptionen bei 295 nm (ε = 1.0 104 l mol-1 cm-1) und 460 nm (ε = 2.5 103 l mol-1 cm-1) für das PhenylthiylRadikal PhS•. Die Banden bei 380 bzw. 570 nm können einem Phenoxyl-Radikal zugeordnet werden. Auffällig ist hierbei die geringe Intensität der Bande bei 380 nm, sowie deren hypsochrome Verschiebung um etwa 30 nm zu den in der Literatur beschriebenen. Der energieärmste Übergang bei 690 nm kann mit einer Metallzu-Ligand-Charge-Transfer-Bande vom Platin zum oxidierten Liganden erklärt werden. Die Cyclovoltammogramme sind vor und nach der Einelektronenoxidation identisch. Die oxidierte Verbindung lässt sich elektrochemisch in ihre reduzierte Form zurückführen. Die Messung des EPR-Spektrums der oxidierten Substanz [Pt(psp)2]+• bei 10 K zeigt ein S=1/2-Signal. Das experimentelle EPR-Spektrum kann mit folgenden Parametern simuliert werden: gz = 2.035 gy = 2.022 gx = 1.992 Wx = 3.3 mT Wy = 3.8 mT Wz = 3.0 mT Azz = 1.5 mT Inuc = 1/2 Komplexe mit dem Liganden Hpsp 69 Sim. Exp. 310 320 330 340 350 360 B [mT] Abb. 6.2.6: EPR-Spektrum von [Pt(psp)2] +• in CH2Cl2; T = 10 K; Leistung: 200 nW; Frequenz: 9.4381 GHz; Modulation: 1.25 mT Die ermittelten g-Werte belegen die ligandenzentrierte Oxidation des Komplexes. Für eine Oxidation des Platin(II) zu Platin(III) erwartet man ein stark anisotropes Signal.[82] Das hier vorliegende leicht axiale Spektrum zeigt jedoch keine deutliche Anisotropie (∆gz-gx = 0.043), wie sie für ein Platin(III)ion in der Literatur beschrieben wird (∆gz-gx = 0.227). Komplexe mit dem Liganden Hpsp 6.3. 70 Der Palladiumkomplex [Pd(psp)2] Der durch Umsetzung von zwei Äquivalenten Hpsp und einen Äquivalent PdCl2 unter Verwendung von zwei Äquivalenten NaOH erhaltene schwach violette Komplex [Pd(psp)2] wurde aus n-Hexan umkristallisiert. Es wurden violette Nadeln erhalten, die sich für eine Röntgenstrukturanalyse eigneten. Die abgebildete Kristallstruktur (Abb. 6.3.1.) zeigt das Palladium(II)ion in einer quadratisch-planaren Koordinationssphäre. cis trans Abb. 6.3.1: Kristallstruktur des Komplexes [Pd(psp)2] In einer Elementarzelle befinden sich zwei cis- und ein trans-Isomer. Beide Isomere sind oben abgebildet. Komplexe mit dem Liganden Hpsp 71 Bindung Bindungslänge [Å] Winkel Bindungswinkel[°] Pd – S(1) 2.2821 (13) S(1)-Pd-O(1) 86.24 (9) Pd – S(2) 2.2872 (14) S(1)-Pd-S(2) 101.64 (5) Pd – O(1) 2.004 (3) O(1)-Pd-O(2) 86.22 (13) Pd – O(2) 1.987 (3) S(1)-Pd-O(2) 169.51 (12) Tab. 6.3.1: Ausgewählte Bindungslängen und –winkel in cis-[Pd(psp)2] Bindung Bindungslänge [Å] Winkel Pd – S(3) 2.2877 (14) S(3)-Pt-O(3) 86.43 (10) Pd – S(3A) 2.2877 (14) S(3)-Pt-S(3A) 180.0 Pd – O(3) 1.980 (3) O(3)-Pt-O(3A) 180.0 Pd – O(3A) 1.980 (3) O(3)-Pt-S(3A) 95.57 (10) Tab. 6.3.2: Ausgewählte Bindungslängen und –winkel in trans-[Pd(psp)2] Bindungswinkel[°] Sowohl für die cis-, als auch für die trans-Konfiguration besteht die schon beim Platinkomplex dieses Liganden besprochene Möglichkeit der Ausbildung zweier Diastereomere, die sich durch die Konfiguration der beiden pyramidal koordinierten Thioether unterscheiden. Bei dem abgebildeten cis-Isomer besitzt das Schwefelatom S(1) S-Konfiguration, wogegen das Schwefelatom S(2) RKonfiguration aufweist. Die Phenylsubstituenten am Schwefel befinden sich auf einer Seite der Koordinationsebene (syn). In Analogie zum Platinkomplex [Pt(psp)2] sind die beiden (R,R)- und (S,S)-Enantiomere (anti) im NMRExperiment nicht unterscheidbar. Beim ebenfalls abgebildeten trans-Isomer weist der Schwefel S(3) S-Konfiguration, der Schwefel (S3A) hingegen RKonfiguration auf. Die Phenylsubstituenten befinden sich auf gegenüberliegenden Seiten der Koordinationsebene (anti). Für die syn-Form existieren auch hier (R,R)- und (S,S)-Enatiomere. Für das cis-Isomer findet man im Kristall die gleiche Konfiguration, die schon beim Platinkomplex [Pt(psp)2] beobachtet wurde. Wiederum wird die πStapelung der Phenylringe Ursache für das Vorliegen der syn-Form beim cisIsomer sein. Komplexe mit dem Liganden Hpsp 72 Bei dem trans-Isomer ist diese Annäherung der Phenylringe nicht möglich. Man beobachtet konsequenterweise die ohne Berücksichtigung der π-Stapelung energetisch günstigere anti-Form. Das 1H-NMR zeigt bei tiefen Temperaturen für die beiden tert-Butylgruppen des Liganden insgesamt acht Signale. Alle vier möglichen Isomere (syn- und anti-Form jeweils des cis- und des trans-Isomers) sind in Lösung im Gleichgewicht vorhanden. Für die Inversion am Schwefel lässt sich auch in diesem Fall über die Methode der Koaleszenztemperaturbestimmung die freie Aktivierungsenthalpie ∆G# bestimmen. 304 K 302 K 300 K 298 K 1,6 1,5 1,4 1,3 1,2 1,1 1,0 δ [ppm] Abb. 6.3.2: 1 H-NMR des Komplexes [Pd(psp)2] in Dimethylformamid-d7 In deuteriertem Dimethylformamid sind die Signale der tert-Butylgruppen des trans-Isomers bei Raumtemperatur (298 K) bereits nicht mehr getrennt. Es handelt sich um die Singuletts bei δ = 1.21 ppm und 1.32 ppm. Komplexe mit dem Liganden Hpsp 73 Die Signale der tert-Butylgruppen der beiden diastereomeren Formen des cisIsomers (δ = 1.16, 1.18, 1.46, 1.48) sind unter diesen Bedingungen noch getrennt. Die Koaleszenztemperatur wird für diese beiden Gruppen zu Tc = 304 K bestimmt. Die Frequenzdifferenz der beiden Signale bei Raumtemperatur beträgt 7 Hz. Hieraus errechnet sich die freie Aktivierungsenthalpie für die Inversion am Schwefel des cis-Isomers zu: ∆G# = 67.5 +/- 1 kJ/mol Um die freie Aktivierungsenthalpie für die Inversion am Schwefel des transIsomers bestimmen zu können, wurde die Probe abgekühlt und die Koaleszenztemperatur bei Auftreten der Trennung der Signale bestimmt. Komplexe mit dem Liganden Hpsp 74 300 K 288 K 283 K 278 K 263 K 1,38 1,36 1,34 1,32 1,30 1,28 1,26 1,24 1,22 1,20 1,18 δ [ppm] Abb. 6.3.3: 1 H-NMR des Komplexes [Pd(psp)2] in Dimethylformamid-d7 Die Frequenzdifferenzen der beiden Signalgruppen beträgt bei 263 K 28.6 Hz für das tieffeldverschobene Signal und 16 Hz für die Signalgruppe um δ = 1.21 ppm. Koaleszenz wird für letzteres Signal bei 283 K beobachtet, für das tieffeldverschobene bei 288 K. So errechnet sich die Inversionsbarriere am Schwefel der trans-Form zu 60.7 kJ/mol bzw. 60.5 kJ/mol. Es wird also die freie Aktivierungsenthalpie für die syn-anti-Isomerisierung des trans-[Pd(psp)2] bestimmt zu ∆G# = 60.6 +/- 1 kJ/mol Die freie Aktivierungsenthalpie der Inversion am Schwefel liegt also für das trans-Isomer des Palladiumkomplexes etwa 6 kJ/mol niedriger als für das cisIsomer. Komplexe mit dem Liganden Hpsp 75 Dieses entspricht den von Boeré und Willis gefundenen Werten für einen ähnlichen Ligandensatz an Palladium(II)[83]. Zurückgeführt wird die niedrigere Inversionsbarriere am Schwefel der trans-Form auf den Einfluss des trans zum Schwefel koordinierten, weniger stark elektronenziehenden Thioether. Die Inversion am Schwefel verläuft über ein planares, dreifach koordiniertes Schwefelatom mit sp2-Hybridisierung und einem freien Elektronenpaar in einem p-Orbital senkrecht zur Koordinationsebene des Metalls. Me S Abb. 6.3.4: Me S Me S Mechanismus der Inversion am Schwefel Dieses hat zur Folge, dass das stärker elektronenziehende Donoratom in transStellung zum Thioether die Inversionsbarriere erhöht.[83] Somit erklärt sich die um 6 kJ/mol höhere freie Aktivierungsenthalpie für das cis-Isomer, bei dem in trans-Position zum Thioether das Phenolat koordiniert ist. Mit Hilfe der HPLC gelingt die Trennung des cis-Isomers von dem transIsomer. Hierzu wird das Isomerengemisch in Hexan/Methanol (1:4) gelöst. Für die chromatographische Trennung wird eine reversed-phase-Säule verwendet. Die beiden Fraktionen werden bei –20°C gesammelt und das Lösungsmittel im Ölpumpenvakuum bei dieser Temperatur entfernt. Die zurückbleibenden Feststoffe enthalten zu ca. 90 % das reine cis- bzw. trans-Isomer. Komplexe mit dem Liganden Hpsp 76 Löst man diese in deuteriertem Dimethylformamid so kann man im NMRExperiment beobachten, wie sich das Gleichgewicht zwischen cis- und transIsomer in Lösung einstellt. k1 cis trans k-1 Die Anfangskonzentrationen seien a für das cis-Isomer und 0 für das transIsomer; die Gleichung für die Kinetik lautet dann: dx =k1 (a − x) − k −1 x dt (Gleichung 6.3.1.) Für den Punkt, an dem die Gleichgewichtskonzentration erreicht ist, ist dx/dt = 0 und x definitionsgemäß xg. Man erhält: k1 (a − x g ) =k −1 x g (Gleichung 6.3.2.) Eingesetzt in Gleichung 6.3.1. ergibt sich dx = (k1 + k −1 )( x g − x) dt (Gleichung 6.3.3.) Nach Integration im Intervall[a;x] für x und [0;t] für t erhält man den Ausdruck xg (Gleichung 6.3.4.) ) =(k1 + k −1 )t = k obs t xg − x Hierbei ist die beobachtete Geschwindigkeitskonstante kobs gleich der Summe ln( aus den Geschwindigkeitskonstanten der Hin- und Rückreaktion. Ein kinetisches Experiment liefert demnach k1 + k-1 = kobs. Dies führt zu einem Zeitgesetz erster Ordnung. Das Verhältnis k1/k-1 erhält man aus der Lage des Gleichgewichts[84]. Berechnung von k1 und k-1 aus k kobs = k1 + k-1 K = [trans]/[cis] = k1/k-1 k1 = K k-1 k-1 = k1/K Komplexe mit dem Liganden Hpsp 77 eingesetzt: kobs = K k-1 + k-1 kobs = k1 + k1/K kobs = k-1 (K +1) kobs = k1 (1 + 1/K) k-1 = kobs/(K+1) k1 = kobs/(1+1/K) (Gleichungen 6.3.5.a/b) Um die kinetischen Parameter der Umwandlung von der cis- und trans-Form des Palladiumkomplexes [Pd(psp)2] bestimmen zu können, werden die isolierten Formen bei unterschiedlichen Temperaturen in deuteriertem Dimethylformamid gelöst und die Einstellung des Gleichgewichts im NMRExperiment verfolgt. Hierzu integriert man die Signale jeweils einer tertButylgruppe in beiden Isomeren. Das Verhälnis der Intensitäten ergibt direkt den prozentualen Anteil beider Isomere in der Lösung. Die Auftragung des Anteils eines Isomers gegen die Zeit zeigt eine Funktion, die die exponentielle Annäherung an die Gleichgewichtskonzentration wiedergibt. Beispielhaft ist die Einstellung des Gleichgewichts bei 253 K ausgehend von dem isolierten trans-Isomer angegeben (Abb. 6.3.5.). Komplexe mit dem Liganden Hpsp 78 80 70 % trans 60 50 40 30 20 10 0 50 100 150 200 250 300 350 t [min] Abb. 6.3.5: Kinetik der cis-trans-Gleichgewichtseinstellung des Komplexes [Pd(psp)2] bei 253 K in Dimethylformamid-d7 Es kann nun entweder eine einfache Exponentialfunktion gesucht werden, die die Messpunkte erklärt, oder aber man trägt die Messpunkte in linearer Form auf (vgl. Gleichung 6.3.4.) und bestimmt kobs direkt aus der Steigung der Geraden (Abb. 6.3.6.). Komplexe mit dem Liganden Hpsp 79 8 ln(x g/(x g -x)) 6 4 2 0 0 50 100 150 200 250 300 350 t [m in] Abb. 6.3.6: linearisierte Auftragung der Kinetik der cis-trans-Gleichgewichtseinstellung des Komplexes [Pd(psp)2] bei 253 K in Dimethylformaid-d7 In dieser Arbeit wurden die kobs-Werte mit der Exponentialgleichung (Gleichung 6.3.6.) aus der nichtlinearen Kurvenanpassung ermittelt. f ( x) =a1 ⋅ e ( − kx ) + a e (Gleichung 6.3.6.) Für t = 0 erhält man die Startkonzentration (ae + a1) des Isomers, für t = ∞ findet sich die Gleichgewichtskonzentration ae. Die beobachtete Geschwindigkeitskonstante k ist abhängig von der Temperatur, bei der sich das Gleichgewicht einstellt. Außerdem findet sich das temperaturabhängige Endgleichgewicht K und somit auch die einzelnen Geschwindigkeitskonstanten k1 und k-1 für die Hin- und Rückreaktion. Die Ergebnisse der Messungen bei Temperaturen zwischen 243 und 300 K sind in Tabelle 6.3.3. zusammengefasst. Komplexe mit dem Liganden Hpsp 80 T [K] K = [trans]/[cis] kobs [s-1] k-1 [s-1] k1 [s-1] 243 0.19 4.56 10-5 3.82 10-5 7.40 10-6 253 0.23 3.33 10-4 2.70 10-4 6.29 10-5 263 0.27 1.73 10-4 1.37 10-4 3.66 10-5 273 0.29 8.5 10-4 6.59 10-4 1.91 10-4 283 0.29 1.1 10-3 8.54 10-4 2.46 10-4 300 0.35 6.27 10-3 4.63 10-3 1.64 10-3 Tab. 6.3.3: Gleichgewichts- und Geschwindigkeitskonstanten der cis-transIsomerisierung des Komplexes [Pd(ptb)2] Mit Hilfe der Gibbs-Helmholtz-Gleichung ∆G# = ∆H#-T∆S# (Gleichung 6.3.7.) lassen sich aus k1 bzw k-1 die Aktivierungsenthalpie ∆H# und die Aktivierungsentropie ∆S# bestimmen. Hierzu wird die Eyring-Gleichung logarithmiert. log k ∆H # 1 ∆S # =10.32 − ⋅ + T 19.1 T 19.1 (Gleichung 6.3.8.) Bei der Auftragung von log k/T gegen 1/T ergibt sich so aus der Steigung der Geraden die Aktivierungsenthalpie und aus dem Achsenabschnitt die Aktivierungsentropie. Abb. 6.3.7. und Abb. 6.3.8. zeigen die Auftragungen Geschwindigkeitskonstanten der Hin- bzw. Rückreaktion (k1 bzw. k-1) für die Komplexe mit dem Liganden Hpsp 81 -5,0 -5,5 log(k 1/T) -6,0 -6,5 -7,0 -7,5 0,0032 0,0034 0,0036 0,0038 0,0040 0,0042 -1 1/T [K ] Abb. 6.3.7: Eyring-Auftragung log(k/T) gegen T für die Hinreaktion -4,5 -5,0 log(k-1/T) -5,5 -6,0 -6,5 -7,0 0,0032 0,0034 0,0036 0,0038 0,0040 0,0042 -1 1/T [K ] Abb. 6.3.8: Eyring-Auftragung log(k/T) gegen T für die Rückreaktion Komplexe mit dem Liganden Hpsp 82 Für die Hinreaktion ergibt sich aus der linearen Regression eine negative Steigung von -2584+/-410 und ein Achsenabschnitt von 3.24+/-1.5. Für die Rückreaktion wird eine negative Steigung von –2275+/-403 und ein ebenfalls positiver Achsenabschnitt von 2.66+/-1.5 ermittelt. In Tab. 6.3.4. werden die sich daraus ergebenen Werte für ∆H# und ∆S# aufgelistet. ∆H# ∆S# Rückreaktion 43.5+/-7.7 kJ/mol -145+/-29 J/(mol K) Hinreaktion 49.4+/-7.8 kJ/mol -136+/-29 J/(mol K) Tab. 6.3.4. ∆H# und ∆S# mit Abweichungen für die cis-transIsomerisierung des Palladiumkomplexes [Pd(psp)2] Da sich das cis- und das trans-Isomer thermodynamisch nicht wesentlich unterscheiden und die Umwandlung der einen in die andere Form über den gleichen Übergangszustand verläuft, sollten die Aktivierungsenthalpien ∆H# und Aktivierungsentropien ∆S# für die Hin- und Rückreaktion nahezu gleich sein. Die in Tab. 6.3.4. zusammengefassten Ergebnisse bestätigen diese Überlegung. Die Differenzen Aktivierungsentropien sollten der den Aktivierungsenthalpien thermodynamischen und Größen Reaktionsenthalpie ∆H0 und Reaktionsentropie ∆S0 entsprechen. Die Berechnung der thermodynamischen Größen erfolgt über die Temperaturabhängigkeit der Gleichgewichtskonstanten K. Mit ∆H 0 ∆S 0 ln K = − + R ⋅T R (Gleichung 6.3.9.) ergibt die Van´t-Hoff-Auftragung ln K gegen 1/T aus der Steigung der Geraden ∆H0 und aus dem Achsenabschnitt ∆S0. Komplexe mit dem Liganden Hpsp 83 -1.0 -1.1 -1.2 ln K -1.3 -1.4 -1.5 -1.6 -1.7 -1.8 0.0032 0.0034 0.0036 0.0038 0.0040 0.0042 -1 1/T [K ] Abb. 6.3.9: Van´t Hoff-Diagramm Es lassen sich folgende Werte ermitteln: Steigung: -720 +/- 88 Achsenabschnitt: 1.36 +/- 0.33 Daraus ergibt sich: ∆H0 = +5.98 +/- 0.73 kJ/mol ∆S0 = +11.3 +/- 2.7 J/(mol K) ∆G0 = +2.6 kJ/mol Für die Bestimmung von ∆S0 ist die Achsenabschnittsbestimmung wenig genau. Mit ∆G0 = - R T ln K läßt sich das ∆G0 für die jeweilige Temperatur bestimmen. Die Auftragung von ∆G0 gegen T liefert eine Gerade mit der Steigung ∆S0. Komplexe mit dem Liganden Hpsp 84 3.4 3.0 0 ∆ G [kJ/mol] 3.2 2.8 2.6 240 250 260 270 280 290 300 T [K] Abb. 6.3.10: Bestimmung von ∆S0 und ∆H0 aus ∆G0 = ∆H0 –T ∆S0 Es ergibt sich: ∆S0 = + 11.0 +/- 2.6 J/(mol K) ∆H0 = + 5.89 +/- 0.72 kJ/mol ∆G0 = + 2.6 kJ/mol Für den Mechanismus der cis-trans-Isomerisierung können zwei unterschiedliche Wege diskutiert werden. Zum einen besteht die Möglichkeit, dass die Umwandlung des einen quadratisch-planaren Isomers in das andere über einen tetraedischen Übergangszustand verläuft. Zum anderen ist ein fünffach koordiniertes Lösungsmittelmoleküls Intermediat denkbar[85]. Abbildung 6.3.11. dargestellt. Beide durch Koordination Mechanismen sind eines in der Komplexe mit dem Liganden Hpsp S 85 S S O O Pd O S Pd O S S Pd O S O O S O Pd LM S S O Pd O O Pd O S S Abb. 6.3.11: Mögliche Mechanismen der Isomerisierung von [Pd(psp)2] Ein ebenfalls denkbarer Dissoziationsprozeß mit einem symetrischen, ebenen, trigonalen Zwischenprodukt Substitutonsreaktionen erscheint quadratischer sehr Komplexe unwahrscheinlich, immer durch weil einen Verdrängungsmechanismus vor sich gehen. Bei diesen Substitutionsreaktionen wird das trigonal-bipyramidale oder quadratisch-pyramidale Intermediat postuliert[86-88]. Komplexe mit dem Liganden Hpsp 86 Um zu klären, welchen Einfluß das Lösungsmittel auf die Geschwindigkeit der Gleichgewichtseinstellung hat, wurde die cis-trans-Isomerisierung, ausgehend von dem isolierten trans-Isomer, bei Raumtemperatur in deuteriertem Toluol durchgeführt. Eine fünffach koordinierte Zwischenstufe kann ausgeschlossen werden, wenn die Gleichgewichtseinstellung nicht signifikant langsamer ist, als das in Dimethylformamid beobachtet wurde. Der Reaktionsverlauf in den unterschiedlichen Lösungsmitteln bei Raumtemperatur ist in den Abbildungen 6.3.12 und 6.3.13 dargestellt. Die Einstellung des Gleichgewichtes erfolgt in Toluol wesentlich langsamer als in dem koordinierenden Lösungsmittel Dimethylformamid. Die erhaltenen Geschwindigkeits- und Gleichgewichtskonstanten sind in Tabelle 6.3.5. gegenübergestellt. K [cis]/[trans] k [s-1] k-1 [s-1] k1 [s-1] DMF-d7 0.35 6.27 10-3 4.63 10-3 1.64 10-3 Toluol-d8 1.22 5.17 10-5 2.33 10-5 2.84 10-5 Tab. 6.3.5. Gleichgewichts- und Geschwindigkeitskonstanten der cis-transIsomerisierung bei 300 K in deuteriertem Dimethylformamid und deuteriertem Toluol Die Gleichgewichtseinstellung vollzieht sich bei gleicher Temperatur in dem nicht koordinierendem Lösungsmittel Toluol hundertmal langsamer als in dem gut koordinierendem Lösungsmittel DMF. Das läßt den Schluss zu, dass der Mechanismus der cis-trans-Isomerisierung über ein fünffach koordiniertes Intermediat verläuft. Für diesen Befund spricht auch, daß bei der Aufarbeitung der mittels HPLC getrennten Isomere bei tiefen Temperaturen gearbeitet werden muß (-20°C). Die Trennung findet in einem Methanol/Hexan-Gemisch statt. Bei der Trennung der Isomere über eine Kieselgelsäule mit einem Hexan/Diethylether-Gemisch als Laufmittel bleiben die getrennten Fraktionen hingegen bei Raumtemperatur relativ stabil. Komplexe mit dem Liganden Hpsp 87 100 % trans 90 80 70 60 50 0 200 400 600 800 1000 1200 t [min] Abb. 6.3.12: Kinetik der cis-trans-Gleichgewichtseinstellung des Komplexes [Pd(psp)2] bei 300 K in Toluol-d8 81 80 79 % cis 78 77 76 75 74 73 0 10 20 30 40 50 60 t [min] Abb. 6.3.13: Kinetik der cis-trans-Gleichgewichtseinstellung des Komplexes [Pd(psp)2] bei 300 K in Dimethylformamid-d7 Komplexe mit dem Liganden Hpsp 88 Dass sich das Gleichgewicht bei Raumtemperatur in Dimethylformamid bei etwa 3/1 (cis/trans) einstellt, in Toluol hingegen bei ziemlich exakt 1/1, ist mit der unterschiedlichen Polarität der beiden Isomeren Formen zu erklären. Das trans-Isomer ist vollständig unpolar, wogegen das cis-Isomer eine gewisse Polarität aufweist. Damit wird das cis-Isomer in polaren Lösungsmitteln durch Solvatationseffekte energetisch abgesenkt, während in Lösungsmitteln (Toluol) kein stabilisierender Effekt erwartet wird. apolaren Komplexe mit dem Liganden Hpsp 89 Das Cyclovoltammogramm des Komplexes [Pd(psp)2] zeigt in Dichlormethan zwei reversible ligandenzentrierte Oxidationen bei +600 und +960 mV gegen Fc/Fc+. 6 4 2 0 I [ µ A] -2 -4 -6 -8 -10 -12 -14 1400 1200 1000 800 600 E [m V] gg. Fc/Fc 400 200 0 + Abb. 6.3.14: Cyclovoltammogramm des Komplexes [Pd(psp)2] bei 20, 50, 100, 200, 400 mV/s; in CH2Cl2 (0.1 M TBAPF6); T = 298 K Im Cyclovoltammogramm des Palladiumkomplexes fällt die zweite Oxidation ungewöhnlich breit aus. Die Vermutung, dass es sich hierbei um zwei Oxidationen mit dicht beieinanderliegenden Potentialen handelt, wird durch die Square-wave-Messung bestätigt. Bei niedrigen Frequenzen wird ersichtlich, dass es sich um zwei Oxidationen bei ähnlichem Potential handelt. Dieses erklärt sich aus dem oben beschriebenen Auftreten eines cis- und eines trans-Isomers bei diesem Komplex. Die beiden Spezies besitzen ähnliche, aber nicht gleiche Oxidationspotentiale. Die Potentiale dieser zweiten und dritten Oxidation liegen bei +920 bzw. +1020 mV gegen Fc/Fc+. Damit entspricht die Potentialdifferenz zwischen der ersten und der zweiten Oxidation 320 mV. Diese Differenz ist identisch mit der Potentialdifferenz, die für den cisPlatinkomplex [Pt(psp)2] gefunden wird. Komplexe mit dem Liganden Hpsp 90 Die zweite Oxidation wird somit dem cis-Isomer, die dritte Oxidation dem trans-Isomer des Palladiumkomplexes [Pd(psp)2] zugeordnet. Ob auch die erste Oxidation der beiden Isomere bei unterschiedlichen Potentialen stattfindet, kann wegen des Auflösungsvermögens der verwendeten elektrochemischen Verfahren nicht geklärt werden. 0 -2 -4 -6 -8 I [µ A] -10 -12 -14 -16 -18 -20 -22 -24 1200 1000 800 600 E [m V] gg. Fc/Fc 400 200 0 + Abb. 6.3.15: Square-wave-Voltammogramm des Komplexes [Pd(psp)2] bei 15, 30, 60, 120 Hz; in CH2Cl2 (0.1 M TBAPF6); T = 298 K Die elektrochemisch durch potentialkontrollierte Coulometrie erzeugte Spezies erwies sich auch bei tiefen Temperaturen als zu instabil, um weitergehende spektroskopische Untersuchungen daran vornehmen zu können. Während der Coulometrie zeigten die parallel durchgeführten Absorptionsspektren schon während dieses Experiments Zersetzung. Komplexe mit dem Liganden Hpsp 6.4. 91 [Pt(psp)2] und [Pd(psp)2] – ein Vergleich Betrachtet man die Ergebnisse der kinetischen Messungen aus den Kapiteln 6.2. und 6.3. so erkennt man trotz großer Ähnlichkeiten der beiden Komplexe [Pt(ptb)2] und [Pd(ptb)2] Tendenzen und Unterschiede, die hier noch einmal zusammenfassend dargestellt werden sollen. Besondere Aufmerksamkeit gilt in diesem Zusammenhang der Tatsache, dass nur beim Palladiumkomplex das cis- und trans-Isomer beobachtet wird, beim Platinkomplex jedoch ausschließlich die cis-Konfiguration auftritt. Außerdem sollen die Koaleszenztemperaturmessungen Unterschiede gewonnenen der Werte aus für die den freien Aktivierungsenthalpien der Inversion am Schwefel diskutiert werden. Beiden Metallen ist eine große Affinität zum Schwefel und eine deutlich geringere zum Sauerstoff gemein. Die Ursache ist im großen Umfang auf die Bildung von Metall-Ligand-π-Bindungen durch Überlappung von besetzten dπOrbitalen (dxz, dxy und dyz) am Metall mit leeren dπ-Orbitalen in der Valenzschale des schweren Donoratoms zurückzuführen. Bei quadratisch-planaren Komplexen, bei denen weiche Donoratome an ein Metall-Atom der Klasse b („weiche“ Zentren) koordinieren, destabilisieren sich zwei weiche Donoren in trans-Stellung zueinander (Antisymbiose)[89]. Dieser Effekt ist ursächlich für die Bevorzugung des cis-Isomers bei beiden Komplexen. Die Tatsache, dass das Platin(II)-Ion noch leichter zu polarisieren (weicher) ist, als das Palladium(II)-Ion und die Pt(II)-Komplexe sich durch eine besonders hohe kinetische Trägheit auszeichnen, erklärt das einzig der cis[Pt(ptb)2]-Komplex beobachtet werden kann. Pd(II)-Komplexe sind sowohl in kinetischer als auch in thermodynamischer Hinsicht weniger stabil als ihre Platin-Homologen. Bei dem [Pd(ptb)2]-Komplex stellt sich daher ein Gleichgewicht zwischen den beiden Isomeren ein. Der Mechanismus dieser Gleichgewichtseinstellung verläuft über ein fünffach koordiniertes Intermediat unter Koordination eines Lösungsmittelmoleküls. Die kinetischen Parameter dieser Umwandlung konnten in Kapitel 6.3. bestimmt werden. Komplexe mit dem Liganden Hpsp 92 Die freien Aktivierungsenthalpien der Umwandlung der diastereomeren synund anti-Formen weisen für die drei untersuchten Komplexe deutliche Unterschiede auf. In Tabelle 6.4.1. sind die Ergebnisse der Koaleszenztemperaturmessungen an dem Palladiumkomplex [Pd(ptb)2] und dem Platinkomplex [Pt(ptb)2] gegenübergestellt. [cis-Pt(ptb)2] [cis-Pd(ptb)2] [trans-Pd(ptb)2] ∆G# [kJ/mol] 77.5 +/-1 67.5 +/-1 60.6 +/-1 Tabelle 6.4.1. freie Aktivierungsenthalpien für die Inversion am Schwefel Boeré und Willis[83] ermittelten für die Inversionsbarrieren am Thioether vergleichbarer Komplexe freie Aktivierungsenthalpien, die 8 – 12 kJ/mol größer sind, jedoch die gleiche Tendenz für die Platin- und Palladiumkomplexe aufweisen. M(II) = Pd, Pt F F O cis-PtL2 : ∆G# [kJ/mol] = 89 S cis-PdL2 : ∆G# [kJ/mol] = 73.5 Ph trans-PdL2 : ∆G# [kJ/mol] = 68 F M(II) 2 Abb. 6.4.1 : Die von Boeré und Willis beschriebenen Komplexe Ursache für die geringere Aktivierungsenergie bei den Palladiumkomplexen gegenüber dem Platinkomplex ist zunächst einmal die geringere Elektronegativität des Palladiums (1.35) im Vergleich zu Platin (1.44)[90]. Eine größere Elektronegativität des Metalls, an das ein Thioether koordiniert ist, erhöht den s-Charakter des verbleibenden nicht bindenden Orbitals am Schwefel. Dies führt zu einer Erhöhung der Inversionsbarriere[91]. Komplexe mit dem Liganden Hpsp 93 Ein weiterer Faktor ist die Stabilisierung des Übergangszustandes, der einen planaren Schwefel aufweist, durch die effizientere π–Überlappung zwischen Schwefel und Palladium (3p-4d) im Vergleich zu der Überlappung zwischen Schwefel und Platin(3d-5d)[83]. Die niedrigere Inversionsbarriere für das trans-Isomer verglichen mit dem cisIsomer des Palladiumkomplexes ist, wie schon in Kapitel 6.3. berichtet, auf die elektronenziehenden Eigenschaften der jeweils trans zum Thioether koordinierten Liganden zurückzuführen. Die Tatsache, dass die Inversionsbarrieren in den hier beschriebenen Komplexen grundsätzlich etwa 10 kJ/mol niedriger liegen, als bei den von Boeré beschriebenen Komplexen, liegt an der unterschiedlichen Elektronendichte am Metall. Während Boeré durch die elektronenziehende CF3-Gruppe die Elektronendichte am Metall herabsetzt, ist bei den in dieser Arbeit untersuchten Komplexen die Elektronendichte am Metall durch die tertButyl substituierten Aromaten deutlich erhöht. Eine höhere Elektronendichte am Metall bedingt eine Herabsetzung der Inversionsbarriere am Schwefel. Ein Vergleich der von Boeré bestimmten Reaktionsenthalpie ∆H° der cistrans-Isomerisierung des Palladiumkomplexes(10.6 kJ/mol), mit der in Kapitel 6.3. ermittelten (11.2 kJ/mol), ergibt eine deutliche Übereinstimmung. Komplexe mit dem Liganden Hbsbu 94 7. Komplexe mit dem Liganden H2bsbu 7.1. Der Platinkomplex [Pt(bsbu)] Der Platinkomplex [Pt(bsbu)] konnte durch Umsetzung des Liganden H2bsbu mit PtCl2 unter Zusatz von zwei Äquivalenten Triethylamin in Acetonitril dargestellt werden. Aus der gelben Lösung fiel hellgelbes [Pt(bsbu)]. Der berechneten Masse von 723.94 g/mol entsprechend findet sich in der Massenspektroskopie der Molekülpeak bei m/z = 723. S S Pt O Abb. 7.1.1: Die Der Komplex [Pt(bsbu)] elektrochemischen wiederum O in entgastem Untersuchungen Dichlormethan. dieses Es Komplexes ergibt sich erfolgen ein dem elektrochemischen Verhalten des Platinkomplexes [Pt(psp)2] vergleichbares Bild (Abb. 7.1.2.). Die Potentiale der beiden reversiblen Oxidationen liegen mit +660 und +925 mV gegen Fc/Fc+ im gleichen Bereich, wie bei dem in Kap. 6.2. besprochenen Platinkomplex [Pt(psp)2]. Während die erste Oxidation bei exakt gleichem Potential stattfindet, liegt das Potential der zweiten Oxidation 65 mV kathodisch gegenüber dem Potential der zweiten Oxidation des vergleichbaren Platinkomplexes aus Kapitel 6.2. verschoben. Aus den Erkenntnissen, die bei der elektrochemischen Oxidation des Platinkomplexes [Pt(psp)2] (Kap. 6.2.) gewonnen werden konnten, lässt sich schlußfolgern, dass es sich auch hier um ligandenzentrierte Oxidationen handelt. Komplexe mit dem Liganden Hbsbu 95 6 4 2 I [µ A] 0 -2 -4 -6 -8 -10 1400 1200 1000 800 600 E [m V] gg. Fc/Fc Abb. 7.1.2: 400 200 0 + Cyclovoltammogramm des Komplexes [Pt(bsbu)] bei 20, 50, 100, 200 und 400 mV/s; in CH2Cl2 (0.1 M TBAPF6); T = 298 K Zur Untersuchung der oxidierten Spezies wurde auch in diesem Fall der Komplex einer potentialkontrollierten Coulometrie unterzogen. Der Komplex kann bei –25°C in entgastem Dichlormethan elektrochemisch oxidiert werden. Die parallel aufgenommenen Elektronenanregungsspektren zeigen ein ähnliches Bild, wie es bei der elektrochemischen Oxidation des Komplexes [Pt(psp)2] beobachtet werden kann. Komplexe mit dem Liganden Hbsbu 96 25 1,6 9 1,4 20 1,2 ε [10 l*mol *cm ] ε [10 l*mol *cm ] 8 7 6 5 4 1,0 0,8 0,6 0,4 0,2 0,0 ε [103 l*mol-1*cm-1] -0,2 15 3 -0,4 260 270 280 290 300 310 320 330 400 λ [nm] 500 600 700 800 900 λ [nm] 10 5 0 200 400 600 800 1000 λ [nm] Abb. 7.1.3: UV-Vis-Spektren während der Coulometrie des Komplexes [Pt(bsbu)] in CH2Cl2 (0.1 M TBAPF6); T = 248 K; c = 2 x 10-4 mol/l Das UV-Vis-Spektrum des nicht oxidierten Komplexes zeigt im sichtbaren Bereich keine Absorptionsmaxima. Im UV-Bereich befinden sich zwei Absorptionen bei 250 (ε = 2.4 104 l mol-1 cm-1) und 325 (ε=4.5 103 l mol-1cm-1) nm, sowie zwei Schultern bei 275 (ε = 8.5 103 l mol-1 cm-1) und 380 (ε = 0.5 103 l mol-1 cm-1) nm. Die Banden unter 350 nm werden π-π* Übergängen des aromatischen Systems zugeordnet, die intensitätsschwache Absorption bei 380 nm einem d-dÜbergang. Während der elektrochemischen Oxidation beobachtet man eine Abnahme der Absorption bei 275 nm und zeitgleich das Auftreten mehrerer Absorptionsbanden im UV- wie auch im sichtbaren Bereich des Spektrums. Komplexe mit dem Liganden Hbsbu 97 Es treten neue Absorptionen bei 300 (ε = 6.5 103 l mol-1 cm-1), 380 (ε = 0.9 103 l mol-1 cm-1), 450 (ε = 0.7 103 l mol-1 cm-1), 570 (ε = 0.5 103 l mol-1 cm-1) und 680 (ε = 0.55 103 l mol-1 cm-1) nm auf. Bei 282 nm findet sich ein isosbestischer Punkt. Die Ähnlichkeit der UV-Vis-Spektren, die während der Coulomterie der beiden Platinkomplexe [Pt(psp)2] und [Pt(bsbu)] aufgenommen wurden, ist offensichtlich. Die Zuordnung der Banden entspricht der Zuordnung, die für den Komplex [Pt(psp)2]+• in Kapitel 6.2. getroffen werden. Die Banden bei 300 und 450 nm werden dem Schwefelradikal zugeordnet, die Banden bei 380 und 570 nm dem Phenoxyl-Radikal. Bei 680 nm findet sich die Metall-zuLigand-Charge-Transfer Bande. Das lässt auch in dem Fall der Oxidation des Komplexes [Pt(bsbu)] eine ligandenzentrierte Oxidation erkennen. -e[Pt(bsbu)]+ [Pt(bsbu)] +e - Bestätigt wird dieses durch die Aufnahme eines S-Band EPR-Spektrums des Komplexes [Pt(bsbu)]+•. Sim. Exp. dX´´ dB 120 122 124 126 128 130 132 134 136 B [mT] Abb. 7.1.4: Experimentelles und simuliertes EPR-Spektrum des Komplexes [Cu2 (bsb)2] in CH2Cl2 T = 52 K; Frequenz: 3.6249 GHz; Leistung: 988.7 µW; Modulation: 0.963 mT Komplexe mit dem Liganden Hbsbu 98 Das experimentelle EPR-Spektrum kann mit folgenden Parametern simuliert werden: gz = 2.06 gy = 2.03 gx = 1.99 Wz = 4.7 mT Wy = 1.1 mT Wx = 1.7 mT Azz = 3.1 mT Inuc = 1/2 Diese Werte stimmen nahezu mit denen des oxidierten Platinkomplexes [Pt(psp)2]+• überein. Wie schon im vorhergehenden Kapitel besprochen belegen auch die hier ermittelten g-Werte die ligandenzentrierte Oxidation des Komplexes. Die Möglichkeit einer metallzentrierten Oxidation kann ausgeschlossen werden. Für eine Oxidation des Platin(II) zu Platin(III) erwartet man ein stark anisotropes Signal.[82] Das hier vorliegende Spektrum zeigt jedoch keine Anisotropie der g-Werte in den Größenordnungen ∆gz-gx = 0.227, wie das für ein Platin(III)ion beschrieben wird. Auch in diesem Fall ist der zweifach oxidierte Komplex nicht stabil. Bei dem Versuch, die zweifach oxidierte Spezies durch eine potentialkontrollierte Coulometrie darzustellen, beobachtet man bei zeitgleich aufgenommenen Elektronenanregungsspektren, wie bei dem Fall des Platinkomplexes [Pt(psp)2]+•, einen Zerfall des Komplexes. Es werden weder isosbestische Punkte noch regelmässigen Bandenveränderungen beobachtet. Es kann also über den Charakter des zweifach oxidierten Komplexes [Pt(bsbu)] auch in diesem Fall keine Aussage gemacht werden. Komplexe mit dem Liganden Hbsbu 7.2. 99 Der Palladiumkomplex [Pd(bsbu)] Die Darstellung des Palladiumkomplexes [Pd(bsbu)] erfolgt durch Umsetzung des Liganden H2bsbu und PdCl2 unter Zusatz von zwei Äquivalenten Triethylamin in Acetonitril. Der erhaltene rötliche Niederschlag wird abfiltriert und gründlich mit Acetonitril und Methanol gewaschen. In der Massenspektroskopie findet sich der Molekülpeak bei m/z = 634. Das entspricht der berechneten Masse des Komplexes [Pd(bsbu)] von 635.28 g/mol. S S Pd O Abb. 7.2.1: O Der Komplex [Pd(bsbu)] Die elektrochemischen Untersuchungen dieses Komplexes in entgastem Dichlormethan zeigen zwei reversible Oxidationen. Aufgrund ihrer Potentiallagen (+600 und +855 mV gegen Fc/Fc+) und der Potentialdifferenz (∆E = 255 mV) müssen die beobachteten Oxidationen, in Analogie zum Platinkomplex [Pt(bsbu)], als ligandenzentriert gedeutet werden. Die beiden Potentiale sind, im Vergleich zu den bei dem Platinanalogen [Pt(bsbu)] beobachteten, um ca. 60 mV kathodisch verschoben. Die Potentialdifferenz ist bei beiden Komplexen jedoch identisch. Komplexe mit dem Liganden Hbsbu 100 Das Cyclovoltammogramm des Komplexes [Pd(bsbu)] ist abgebildet (Abb. 7.2.2.). 8 6 4 2 I [µ A] 0 -2 -4 -6 -8 -10 -12 -14 1400 1200 1000 800 600 E [m V] gg. Fc/Fc Abb. 7.2.2: 400 200 0 + Cyclovoltammogramm des Komplexes [Pd(bsbu)] bei 50, 100, 200 mV/s; in CH2Cl2 (0.1 M TBAPF6); T = 298 K [Pd(bsbu)] [Pd(bsbu)]+ Der Komplex [Pd(bsbu)]+• ist auch bei tiefen Temperaturen nicht stabil. Schon während der elektrochemischen Oxidation bei –25°C zeigen die parallel aufgenommenen Elektronenanregungsspektren eine Zersetzung des oxidierten Komplexes. Eine weitergehende Untersuchung dieser Spezies war demnach nicht möglich. Ob sich tatsächlich mit der zweiten Oxidation das Diradikal bildet, oder die entsprechende chinoide Struktur, kann auch hier aufgrund der Instabilität der oxidierten Komplexe nicht geklärt werden. Komplexe mit dem Liganden Hbsbu 7.3. 101 Der Kupferkomplex [Cu(bsbu)] Der Kupferkomplexes [Cu(bsbu)] wurde durch die Umsetzung des Liganden H2bsbu mit Cu(NO3)2 x 3 H2O unter Zusatz von zwei Äquivalenten methanolischer NaOH-Lösung in Methanol erhalten. Der entstandene braune Niederschlag wurde abfiltriert und aus Dichlormethan umkristallisiert. Die berechnete Masse des Komplexes [Cu(bsbu)] von 592.41 g/mol stimmt mit dem in der Massenspektroskopie gefundenen Molekülpeak m/z = 591 überein. S S Cu O Abb. 7.3.1: O Der Komplex [Cu(bsbu)] Das bei 10 K in Dichlormethan aufgenommene X-Band-EPR-Spektrum zeigt ein axiales S = ½ - Signal bei giso = 2.093. Simuliert werden kann das in Abbildung 7.3.2. dargestellte Spektrum mit folgenden Parametern: S=½ Inuc = 3/2 gz = 2.136 gy = gx = 2.071 Azz = 11.27 mT W = 33.3 mT Komplexe mit dem Liganden Hbsbu 102 Das hier beobachtete axiale S = ½ - Signal beweist, das es sich in diesem Fall um eine monomere Spezies handelt. Die Erweiterung der Brücke zwischen den beiden Schwefelatomen erlaubt also wie vermutet eine quadratisch-planare Koordination des Liganden an das Kupferzentrum. Sim. Exp. dX´´ dt 260 280 300 320 340 360 B [mT] Abb. 7.3.2: Experimentelles und simuliertes EPR-Spektrum des Komplexes [Pt(bsbu)] in CH2Cl2 T = 10 K; Frequenz: 9.43 GHz; Leistung: 0.20117 µW; Modulation: 1.25 mT Das Cyclovoltammogramm des Komplexes [Cu(bsbu)] zeigt zwei irreversible Oxidationen bei +565 und +945 mV gegen Fc/Fc+ (Abb. 7.3.3.). Diese Potentiale entsprechen den für den Komplex [Cu2(bsb)2] in Kapitel 5.1. gefundenen. Beide Potentiale sind bei dem Komplex [Cu(bsbu)] lediglich um etwa 50 mV kathodisch verschoben. Auch bei diesem Kupferkomplex waren die oxidierten Substanzen einer spektroskopischen Untersuchung aufgrund hoher Instabilität nicht zugänglich. Komplexe mit dem Liganden Hbsbu 103 4 2 0 -2 I [µ A] -4 -6 -8 -10 -12 -14 -16 1200 1000 800 600 E [m V] gg. Fc/Fc Abb. 7.3.3: 400 200 0 + Cyclovoltammogramm des Komplexes [Cu(bsbu)] bei 50, 100, 200, 400 mV/s; in CH2Cl2 (0.1 M TBAPF6); T = 298 K; Zusammenfassung 8. 104 Zusammenfassung Die Untersuchungen an den Übergangsmetallkomplexen der hier vorgestellten Thioether-Phenolatliganden erbrachten wertvolle Erkenntnisse über deren elektrochemische Eigenschaften und die magnetischen Eigenschaften der synthetisierten Kupferdimere. Sowohl die Strukturen dreier Liganden als auch die Strukturen verschiedener Übergangsmetallkomplexe konnten röntgenstrukturanalytisch bestimmt werden. Es gelang die kinetischen und thermodynamischen Parameter der cis-trans-Isomerisierung zweizähniger Liganden an Palladium zu ermitteln, sowie Einblicke in den Mechanismus dieser Gleichgewichtseinstellung zu gewinnen. Zunächst wurde verschiedener, ein allgemeiner neuartiger Syntheseweg für Thioether-Phenolatliganden die Darstellung entwickelt. Die erfolgreiche Umsetzung mit unterschiedlichen Übergangsmetallen erlaubte systematische Untersuchungen zu Struktur und Eigenschaften der entsprechenden Übergangsmetallkomplexe. In der Abbildung 8.1. sind die im Rahmen dieser Arbeit dargestellten Liganden zusammengefasst. Zusammenfassung 105 S S S S OH OH HO (H2bse) HO (H2bsb) 1,2-Bis (3,5-di-tert-butyl-2- 1,2-Bis (3,5-di-tert-butyl-2- hydroxy-phenylsulfanyl)-ethan hydroxy-phenylsulfanyl)benzen S S O OH H S HO (H2bsbu) (Hpsp) 4,6-Di-tert-butyl-2- 1,2-Bis (3,5-di-tert-butyl-2- phenylsulfanyl-phenol hydroxy-phenylsulfanyl)-butan Abb. 8.1: Synthetisierte Liganden Mit Ausnahme der Komplexe des Liganden H2bsbu konnten sämtliche, in dieser Arbeit vorgestellten, Komplexe durch die Röntgenstrukturanalyse bestätigt werden. Von primärem Interesse war die Darstellung der entsprechenden Kupferkomplexe, die in allen vier Fällen erfolgreich war. Obschon keiner dieser Komplexe katalytisch, im Sinne eines funktionalen Modells der Galaktose Oxidase, aktiv ist, weisen sie einige interessante Eigenschaften auf. Zusammenfassung 106 Bei den Komplexen [Cu2(bse)2] und [Cu2(bsb)2] ließen sich ungewöhnlich starke, ferromagnetische Kopplungen zwischen den beiden Kupferzentren nachweisen. Mit Hilfe einer Analyse der aus der Röntgenstrukturanalyse erhaltenen Strukturparametern konnten diese magnetischen Eigenschaften erklärt werden. Ebenfalls gelang die Synthese der entsprechenden monomeren Spezies. Im Falle des [Cu(bsb)(py)2] wurde die Entstehung der monomeren Spezies durch die Röntgenstrukturanalyse nachgewiesen. Es konnte gezeigt werden, daß die bevorzugte dimere, phenolatüberbrückte Form der Kupferkomplexe bei den Komplexen [Cu2(bse)2] und [Cu2(bsb)2] durch eine Erweiterung der Brücke zwischen den Schwefeldonoren (H2bsbu) oder aber durch den Einsatz eines zweizähnigen Liganden (Hpsp) vermieden werden kann. Die Untersuchungen an den dargestellten Platin- und Palladiumkomplexen des zweizähnigen Liganden Hpsp lieferten eine Vielzahl interessanter Ergebnisse. Es gelang sowohl den Komplex [cis-Pt(psp)2] als auch das cis- und transIsomer des Palladiumkomplexes [Pd(psp)2] röntgenstrukturanalytisch zu bestimmen. Dieses sind die ersten Röntgenstrukturbeispiele einer derartigen Koordinationssphäre an Platin und Palladium. Es wurden hiermit die spektroskopischen Befunde von Boeré und Willis[83] bestätigt, die für einen ähnlichen Ligandensatz aus NMR-Experimenten schlußfolgerten, daß es sich bei dem Platin-Komplex um das cis-Isomer handelt und der Palladiumkomplex in beiden isomeren Formen vorliegt. Durch die pyramidale Geometrie am koordinierenden Schwefel entstehen bei diesen Komplexen pro Isomer zwei Diastereomere, bei denen sich die Phenylringe der Thioether auf der gleichen Seite der Koordinationssphäre befinden (syn) oder auf der jeweils gegenüberliegenden (anti). Durch Koaleszenztemperaturmessungen konnten für alle drei Isomere die Inversionsbarrieren der Umwandlung dieser Diastereomeren ineinander bestimmt werden. Zusammenfassung Auch gelang 107 die Trennung des und cis- trans-Isomeren des Palladiumkomplexes (Pd(psp)2] mittels der HPLC. Erstmals wurden durch eine Reihe kinetischer Messungen die thermodynamischen und kinetischen Größen einer solchen Umwandlung bei einem derartigen Ligandensatz bestimmt. Durch die Variation des Lösungsmittels während der Umwandlung konnten wertvolle Informationen über den Mechanismus dieser Geichgewichtseinstellung gewonnen werden. Die elektrochemischen Eigenschaften der in dieser Arbeit vorgestellten Komplexe wurden mit Hilfe der Cyclovoltammetrie untersucht. Bei den Komplexen Untersuchung [Co(bsb)(Hbsb)], der potentialkontrollierten oxidierten [Pt(psp)2] Spezies. Coulometrie und [Pt(bsbu)] Sowohl die aufgenommenen gelang eine während der Elektronen- anregungsspektren, als auch die an den oxidierten Komplexen durchgeführten EPR-Experimente beweisen, daß es sich um radikaltragende Spezies handelt. Apparativer Teil 9. 108 Apparativer Teil Alle Analysen und Messungen wurden, falls nicht anders vermerkt, am MaxPlanck-Institut für Strahlenchemie in Mülheim an der Ruhr durchgeführt. Elementaranalysen Die Elementaranalysen wurden im mikroanalytischen Labor Kolbe, Mülheim an der Ruhr, durchgeführt. Infrarotspektren Die Infrarotspektren wurden auf einem Perkin-Elmer FT-IR-Spektrometer des Typs 2000 am Max-Planck-Institut für Strahlenchemie in Mülheim an der Ruhr aufgenommen. Die Proben wurden dazu mit Kaliumbromid verrieben und bei Raumtemperatur als Pressling vermessen. Elektronenanregungsspektren Die Aufnahme der Elektronenanregungsspektren erfolgte auf einem Hewlett Packard Dioden-Array-Spektrometer des Typs HP8452A im Wave Voltammogramme und Wellenlängenbereich von 190 bis 1100 nm. Elektrochemische Experimente Die Cyclovoltammogramme, Square Coulometrien wurden am Max-Planck-Institut für Strahlenchemie in Mülheim an der Ruhr auf einem Gerät der Firma EG&G (Potentiostat/Galvanostat Model 273A) bei verschiedenen Temperaturen mit einer von E. Bothe, P. Höfer, R. Höfer entwickelten Messapparatur aufgenommen. Die Drei-ElektrodenMeßzelle bestand aus einem Silberdraht als Gegenelektrode (GE), einer GlasKohlenstoff-Scheibenelektrode (GC) als Arbeitselektrode (AE) und einer Ag//0.01 M AgNO3/CH3CN Referenzelektrode (RE). Als Leitsalzlösung wurde Apparativer Teil 109 eine 0.1 mol/l Lösung von Tetra-n-butylammoniumhexafluorophophat in wasserfreiem Dichlormethan verwendet. Als interner Standart wurde nach der Messung der zuvor untersuchten Lösung Ferrocen zugesetzt, dessen Potential (Fc/Fc+) unabhängig vom Lösungsmittel +0.4 V gegen die Normalwasserstoffelektrode (NHE) beträgt.[92] Für die Coulometrien wurde eine Platinnetz-Arbeitelektrode verwendet. Massenspektren Die EI-Massenspektren wurden in der Arbeitsgruppe von Herrn D. Stöckigt am Max-Planck-Institut für Kohlenforschung in Mühlheim an der Ruhr durchgeführt. Messungen der magnetischen Suszeptibilität Die Magnetmessungen wurden von Herrn A. Göbels am Max-Planck-Institut für Strahlenchemie in Mülheim an der Ruhr auf einem SQUID-Magnetometer MPMS der Firma Quantum Design durchgeführt. Die Proben wurden in einer nahezu kugelförmigen Gelatinekapsel vermessen. Die Responsefunktion wurde bei jeder angegebenen Temperatur vier mal auf einer Länge von 6 cm an 32 Messpunkten gemessen. Die aus der Responsefunktion gefittete Volumenmagnetisierung wurde um den diamagnetischen Anteil des Halters und der Kapsel korrigiert und in die Volumensuszeptibilität umgerechnet. Die so erhaltene Volumensuszeptibilität wurde im Magnetprogramm JULIUS-F1 benutzt. EPR-Spektren Die Messungen der EPR-Spektren bei Raumtemperatur mit Argon begasten Lösungen wurden von K. Hildenbrand und H. Niehaus am Max-Planck-Institut für Strahlenchemie in Mülheim an der Ruhr auf einem modifizierten EPRSpektrometer der Firma Varian in einer Quarzzelle (d = 0.3 mm) durchgeführt. Die Daten wurden mit einer Datenstation der Firma Stelar s.n.c. digitalisiert. Apparativer Teil 110 Die EPR-Spektren bei tiefen Temperaturen wurden von Herrn F. Reikowski am Max-Planck-Institut für Strahlenchemie in Mülheim an der Ruhr mit einem Gerät „ESP 300“ der Firma Bruker aufgenommen, das mit der Mikrowellenbrücke ER 041 XK-D der Firma Bruker und mit einem EPRKryostaten 910 der Firma Oxford Instruments ausgestattet ist. Die Simulation der EPR-Spektren bei Raumtemperatur erfolgte mit dem Computerprogramm „EPR“ von F. Neese in der Version 1.0 von 1994[93]. Die Simulation der EPR-Spektren bei tiefen Temperaturen erfolgte mit dem Programmpaket „ESIM“ von E. Bill[94]. Die Pulversimulation wird über ein Dreiecks-Winkelnetz ausgeführt. Nullfeldaufspaltungen werden mit D und E parametisiert. Die Programme basieren auf der Routine IRON_HS.[95] Insbesondere die numerischen Verfahren für die Suche der Resonanzübergänge auf der B-Feldachse und deren Verfeinerung mittels Newton–RaphsonVerfahren sind von IRON_HS übernommen, ebenso wie die Methode der Pulversummation über ein Dreiecks-Winkelnetz (θ,φ). Die ESIM-Routinen erlauben die automatische Optimierung aller Parameter per SIMPLEXVerfahren.[96] NMR-Spektren Die NMR-Spektren wurden in der Arbeitsgruppe von Herrn N. Metzler-Nolte am Max-Planck-Institut für Strahlenchemie in Mülheim an der Ruhr gemessen. Die Messungen erfolgten auf NMR-Spektrometern des Typs ARX 250(1H bei 250.13 MHz und 13C), DRX 400 (1H bei 400.13 MHz und 13C) und DRX 500 (1H bei 500.13 MHz und 13C) der Firma Bruker. Die Angabe der 1H- und 13CVerschiebung erfolgte gegen die Referenz TMS unter Benutzung der Signale nicht deuterierter Lösungsmittelmoleküle als inneren Standart. Röntgenstrukturanalyse Die Röntgenstrukturanalysen wurden von Herrn T. Weyhermüller und Frau H. Schucht auf einem Enraf-Nonius CAD4, Nonius Kappa-CCD oder einem Siemens SMART System durchgeführt (Mo-Kα-Strahlung). Zur Auswertung diente das Programm SHELXTL PLUS. Apparativer Teil 111 HPLC Die Isolierung der cis- und trans-Isomeren des Komplexes [Pd(psp)2] erfolgte am Max-Planck-Institut für Strahlenchemie in Mülheim an der Ruhr auf einer reversed-phase-Säule 250 x 10 mm Nucleosil-100-7-C18 der Firma Macherey und Nagel. Die Apparatur bestand außerdem aus der Steuereinheit SCL-10A, der Pumpe LC-10AD und dem UV-Vis-Detektor SPD-10AV, allesamt von der Firma Shimadzu. Präparativer Teil 10. 112 Präparativer Teil Darstellung von 2-Brom-4,6-di-tert-butylphenol OH Br 51.58 g (0.25 mol) 2,4-di-tert-butylphenol werden in 80 ml CHCl3 gelöst und bei Raumtemperatur 39.95 g (0.25 mol) Brom unter Rühren zugetropft. Das entstehende HBr wird durch eine 10%ige NaOH-Lösung geleitet. Nach 2 Stunden rühren wird die Lösung nacheinander mit einer 10%igen NaHSO3-Lösung, einer 10 %igen NaOH-Lösung und destilliertem Wasser gewaschen. Die wässrige Phase wird dreimal mit Diethylether ausgeschüttelt und die vereinigte organische Phase mit Magnesiumsulfat getrocknet. Das Lösungsmittel wird am Rotationsverdampfer entfernt und das gelbe Öl mit wenig Heptan versetzt. Das Produkt kristallisiert bei -20 °C aus, wird abfiltriert und mit wenig kaltem Heptan gewaschen. Ausbeute: 60.0 g (84%) Molmasse: 285.23 g mol-1 C14H21OBr Kernresonanzspektren: 1 H-NMR (CDCl3, 400 MHz): δ 1.27 (9H, s); 1.39 (9H, s); 5.63 (1H, s); 7.23 (1H, d); 7.31 (1H, d) ppm 13 C-NMR (CDCl3, 100 MHz): δ 29.41; 31.46; 34.41; 35.56; 111.89; 123.68; 126.28; 136.74; 143.71;147.99 ppm Präparativer Teil 113 Darstellung von Benzensulfenylchlorid: SCl Zu einer Lösung von 22.24 g (0.2 mol) Thiophenol in 20 ml getrocknetem Dichlormethan werden bei 0°C unter Rühren und Feuchtigkeitsausschluss 13.5 g (0.1 mol) Sulfurylchlorid zugetropft. Die Reaktion ist beendet, wenn die zunächst auftretende Trübung verschwunden ist und eine orangefarbene, klare Lösung vorliegt. Das Lösungsmittel wird im Ölpumpenvakuum entfernt und das zurückbleibende rötliche Öl bei 35°C und 2 10-2 mbar destilliert. Molmasse: 144.63 g mol-1 C6H5SCl Darstellung von Benzen-1,2-disulfenylchlorid: Zu einer Lösung von 1.42 mg (10 mmol) Benzen-1,2dithiol in 20 ml getrocknetem Dichlormethan werden bei Cl S SCl -20°C unter Rühren und Feuchtigkeitsausschluß 2.70 g (20 mmol) Sulfurylchlorid zugetropft. Die Reaktion ist beendet, wenn die zunächst auftretende Trübung verschwunden ist und eine orange, klare Lösung vorliegt. Das Lösungsmittel wird unter Eiskühlung im Ölpumpenvakuum entfernt und der zurückbleibende Feststoff bei -20°C verwahrt. Die hohe Instabilität verhindert eine Charakterisierung des Produktes. Das Produkt der Umsetzung mit dem metallierten Aromaten belegt jedoch die Entstehung des Sulfenyl Chlorids. Molmasse: 214.14 g mol-1 C6H4S2Cl2 Präparativer Teil 114 Darstellung von Ethan-1,2-disulfenylchlorid: Cl S SCl Zu einer Lösung von 942 mg (10 mmol) 1,2-Ethandithiol in 20 ml getrocknetem Dichlormethan werden bei Raumtemperatur unter Rühren und Feuchtigkeitsausschluss 2.70 g (20 mmol) Sulfurylchlorid zugetropft. Die Reaktion ist beendet, wenn die zunächst auftretende Trübung verschwunden ist und eine gelbliche, klare Lösung vorliegt. Das Lösungsmittel wird unter Eiskühlung im Ölpumpenvakuum entfernt und der zurückbleibende gelbe Feststoff bei -20°C verwahrt. Molmasse: 163.09 g mol-1 C2H4S2Cl2 Kernresonanzspektren: 1 H-NMR (CDCl3, 400 MHz): δ 3.51 (4H, s); Präparativer Teil 115 Darstellung von Butan-1,4-dithiocyanat: In einem 500 ml Dreihalskolben mit KPG-Rührer, Tropftrichter NC S S CN und Rückflusskühler werden 38.0 g (0.5 mol) Ammoniumrhodanid in 120 ml Ethanol vorgelegt und zum Rückfluss erhitzt. In der Siedehitze werden 54.0 g (0.25 mol) 1,4-Dibrombutan langsam zugetropft. Nach 12 Stunden kochen unter Rückfluss wird die Reaktionslösung auf Raumtemperatur abgekühlt und der entstandene weiße Niederschlag abfiltriert. Das Lösungsmittel wird am Rotationsverdampfer entfernt, das zurückbleibende Öl mit 150 ml H2O versetzt und die wässrige Phase dreimal mit Diethylether ausgeschüttelt. Die organische Phase wird über Magnesiumsulfat getrocknet und das Lösungsmittel erneut am Rotationsverdampfer entfernt. Das zurückbleibende gelbe Öl bedarf keiner weiteren Aufarbeitung. Das Produkt ist so in über 95%iger Reinheit zu erhalten. Ausbeute: 34.4 g (89%) Molmasse: 172.16 g mol-1 Massenspektrum: 171 m/z [C6H8S2N2]+ Kernresonanzspektren: 1 H-NMR (CDCl3, 400 MHz): δ 1.96 (4H, m); 2.95 (4H, m) ppm 13 C-NMR (CDCl3, 100 MHz): δ 27.89; 32.86; 111.56 ppm C6H8S2N2 Präparativer Teil 116 Darstellung von 1,2-Dithian: S 34.4 g (0.2 mol) Butan-1,4-dithiocyanat werden unter kurzer S Erwärmung in 80 ml Methanol gelöst und unter Rühren und Kühlung im Eisbad eine Lösung von 24 g (0.4 mol) Kaliumhydroxid in 120 ml Methanol langsam zugefügt. Anschließend wird noch 30 Minuten bei 40°C gerührt. Der ausgefallene Feststoff wird abgesaugt und das Filtrat auf 150 ml Eiswasser gegeben. Das ausfallende Produkt wird abgesaugt und das Filtrat bei 0°C verwahrt. Über Nacht fällt weiteres Produkt aus dem Filtrat. Das Produkt wird mit über 99.5%iger Reinheit erhalten. Ausbeute: 20.0 g (84%) Molmasse: 120.24 g mol-1 C4H8S2 Massenspektrum: 120 m/z [C4H8S2]+ Kernresonanzspektren: 1 H-NMR (CDCl3, 250 MHz): δ 1.94 (4H, m); 2.82 (4H, m) ppm 13 C-NMR (CDCl3, 100 MHz): δ 27.76; 33.31 ppm Darstellung von Butan-1,4-disulfenylchlorid: Zu einer Lösung von 1.2 g (10 mmol) 1,2-Dithian in 20 ml getrocknetem Diethylether werden bei CL S S Cl –20°C unter Rühren und Feuchtigkeitsausschluss 1.35 g (10 mmol) Sulfurylchlorid zugetropft. Die gelbe Reaktionslösung wird langsam auf 0°C erwärmt und sodann bei –20°C maximal 3 Stunden verwahrt. Molmasse: 191.15 g mol-1 C4H8S2Cl2 Präparativer Teil 117 Darstellung von 4,6-Di-tert-butyl-6-phenylsulfanyl-phenol (Hpsp) S 5.71 g (20 mmol) 2-Brom-4,6-di-tert-butylphenol O werden in 30 ml getrocknetem Diethylether in einer ausgeheizten Apparatur unter H Argon vorgelegt und bei -50°C 25 ml einer 1.6 molaren Buthyllithium-Lösung zugetropft. Bei langsamen Erwärmen bis auf Raumtemperatur löst sich der ausfallende Feststoff wieder. Die hellgelbe Lösung wird noch ca. 30 Minuten bei Raumtemperatur gerührt. Nach Abkühlen der Lösung auf -65°C wird langsam eine Lösung von 2.89 g (20 mmol) Benzensulfenyl Chlorid in 10 ml getrocknetem Diethylether zugetropft. Die Lösung wird langsam auf Raumtemperatur gebracht, wobei ein weißer Feststoff ausfällt. Der Reaktionsansatz wird über Nacht weitergerührt. Dann wird vorsichtig mit ca. 20 ml destilliertem Wasser hydrolisiert, die Phasen getrennt und die wäßrige Phase dreimal mit ca. 20 ml Diethylether ausgeschüttelt. Die vereinigte organische Phase wird über Magnesiumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Das Produkt wird bei 1*10-2 mbar und 115°C destilliert. Molmasse: 314.49 g mol-1 Ausbeute: 3.40g (54 %) C20H26OS Elementaranalyse: C% H% S% berechnet: 76.38 8.33 10.20 gefunden: 75.92 8.65 10.01 Massenspektrum: 314 m/z [Hpsp]+ Kernresonanzspektren: 1 H-NMR (CDCl3, 250 MHz): δ 1.28 (9H, s); 1.40 (9H, s); 6.83 (1H, s); 7.00 - 7.25(7H, m) 13 C-NMR (CDCl3, 63 MHz): δ 29.41; 31.49; 34.36; 35.27; 11.57; 125.71; 126.30; 126.82; 129.07;131.03; 135.73; 136.43; 142.77; 153.39 ppm Präparativer Teil Darstellung 118 von 1,2-Bis (3,5-di-tert- butyl-2-hydroxy-phenylsulfanyl)-benzen (H2bsb) S OH S HO 5.71 g (20 mmol) 2-Brom-4,6-di-tertbutylphenol werden in 30 ml getrocknetem Diethylether in einer ausgeheizten Apparatur unter Argon vorgelegt und bei –50°C 25 ml einer 1.6 molaren Buthyllithium-Lösung zugetropft. Bei langsamen Erwärmen bis auf Raumtemperatur löst sich der ausfallende Feststoff wieder. Die hellgelbe Lösung wird noch ca. 30 Minuten bei Raumtemperatur gerührt. Nach Abkühlen der Lösung auf -65°C wird langsam eine Lösung von 2.11 g (10 mmol) Benzen-1,2-disulfenyl Chlorid in 10 ml getrocknetem Diethylether zugetropft. Die Lösung wird langsam auf Raumtemperatur gebracht, wobei ein weißer Feststoff ausfällt. Der Reaktionsansatz wird über Nacht weitergerührt. Dann wird vorsichtig mit ca. 20 ml destilliertem Wasser hydrolisiert, die Phasen getrennt und die wäßrige Phase zweimal mit ca. 20 ml Diethylether und zweimal mit Dichlormethan ausgeschüttelt. Die vereinigte organische Phase wird über Magnesiumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Der zurückbleibende weiße Feststoff wird in Aceton aufgenommen. Das Produkt kristallisiert bei -20°C aus. Molmasse: 550.87 g mol-1 Ausbeute: 3.45g (63 %) C34H46O2S2 Elementaranalyse: C% H% S% berechnet: 74.13 8.42 11.64 gefunden: 74.52 8.53 11.41 Massenspektrum: 550 m/z [H2bsb]+ Kernresonanzspektren: 1 H-NMR (CDCl3, 400 MHz): δ 1.30 (18H, s); 1.40 (18H, s); 6.77 (2H, q); 6.83 (2H, s); 6.97 (2H, q); 7.41 (2H, d) 7.43 (2H, d) ppm 13 C-NMR (CDCl3, 100 MHz): δ 29.42; 31.51; 34.42; 35.34; 115.82; 126.97; 126.99; 127.75; 130.91;135.02; 136.03; 143.01; 153.45 ppm Präparativer Teil 119 Darstellung von 1,2-Bis (3,5-di-tert-butyl-2-hydroxy-phenylsulfanyl)-ethan (H2bse) S 5.71 g (20 mmol) 2-Brom-4,6-di-tertbutylphenol werden in 30 ml getrocknetem Diethylether in einer S OH HO ausgeheizten Apparatur unter Argon vorgelegt und bei -50°C 25 ml einer 1.6 molaren Buthyllithium-Lösung zugetropft. Bei langsamen Erwärmen bis auf Raumtemperatur löst sich der ausfallende Feststoff wieder. Die hellgelbe Lösung wird noch ca. 30 Minuten bei Raumtemperatur gerührt. Nach Abkühlen der Lösung auf -65°C wird langsam eine Lösung von 1.63 g (10 mmol) Ethan-1,2-disulfenyl Chlorid in 10 ml getrocknetem Diethylether zugetropft. Die Lösung wird langsam auf Raumtemperatur gebracht, wobei ein weißer Feststoff ausfällt. Der Reaktionsansatz wird über Nacht weitergerührt. Dann wird vorsichtig mit ca. 20 ml destilliertem Wasser hydrolisiert, eventuell nicht gelöster Feststoff abfiltriert (Produkt !), die Phasen getrennt und die wäßrige Phase zweimal mit ca. 20 ml Diethylether und zweimal mit Dichlormethan ausgeschüttelt. Die vereinigte organische Phase wird über Magnesiumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Der zurückbleibende weiße Feststoff wird in Pentan aufgenommen. Das Produkt kristallisiert bei -20°C aus. Molmasse: 502.83 g mol-1 Ausbeute: 3.62g (72 %) C30H46O2S2 Elementaranalyse: C% H% S% berechnet: 71.66 9.22 12.75 gefunden: 71.89 9.54 12.48 Massenspektrum: 502 m/z [H2bse]+ Kernresonanzspektren: 1 H-NMR (CDCl3, 400 MHz): δ 1.25 (18H, s); 1.37 (18H, s); 2.78 (4H, s); 7.03 (2H, s); 7.28 (2H, d); 7.30 (2H, d) ppm 13 C-NMR (CDCl3, 100 MHz): δ 29.42; 31.51; 34.29; 35.20; 36.24; 117.44; 126.10; 130.30; 135.31; 142.35;153.18 ppm Präparativer Teil 120 Darstellung von 1,2-Bis (3,5-di-tert-butyl-2-hydroxy-phenylsulfanyl)-butan (H2bsbu) 5.71 g (20 mmol) 2-Brom-4,6-di-tert- S butylphenol O werden in 30 ml S H H O getrocknetem Diethylether in einer ausgeheizten Apparatur unter Argon vorgelegt und bei -50°C 25 ml einer 1.6 molaren Buthyllithium-Lösung zugetropft. Bei langsamen Erwärmen bis auf Raumtemperatur löst sich der ausfallende Feststoff wieder. Die hellgelbe Lösung wird noch ca. 30 Minuten bei Raumtemperatur gerührt. Nach Abkühlen der Lösung auf -80°C wird langsam eine Lösung von 1.91 g (10 mmol) Butan-1,4-disulfenyl Chlorid in 10 ml getrocknetem Diethylether zugetropft. Die Lösung wird langsam auf Raumtemperatur gebracht, wobei ein weißer Feststoff ausfällt. Der Reaktionsansatz wird über Nacht weitergerührt. Dann wird vorsichtig mit ca. 20 ml destilliertem Wasser hydrolisiert, die Phasen getrennt und die wäßrige Phase zweimal mit ca. 20 ml Diethylether und zweimal mit Dichlormethan ausgeschüttelt. Die vereinigte organische Phase wird über Magnesiumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Das zurückbleibende gelbe Öl wird aus Methanol umkristallisiert. Molmasse: 530.88 g mol-1 Ausbeute: 2.17g (41 %) C32H50O2S2 Elementaranalyse: C% H% S% berechnet: 72.40 9.49 12.08 gefunden: 70.21 8.94 11.67 Massenspektrum: 530 m/z [H2bsbu]+ Kernresonanzspektren: 1 H-NMR (CDCl3, 250 MHz): δ 1.26 (18H, s); 1.38 (18H, s); 1.66 (4H, p); 2.63 (4H, m); 7.24 (2H, s); 7.27 (2H, d); 7.31 (2H, d) ppm 13 C-NMR (CDCl3, 63 MHz): δ 28.44; 29.39; 31.50; 34.25; 35.16; 36.35; 118.56; 125.69; 130.19; 135.00; 142.07; 152.94 ppm Präparativer Teil 121 Darstellung von 1,2-Bis (4,6-di-tert-butyl-phenol-2-thio)-ethan (H2bpte) S 5.71 g (20 mmol) 2-Brom-4,6-di-tert.butylphenol werden in 30 ml getrocknetem Diethylether in einer S OH HO ausgeheizten Apparatur unter Argon vorgelegt und bei -50 °C 25 ml einer 1.6 molaren Buthyllithium-Lösung zugetropft. Bei langsamen Erwärmen bis auf Raumtemperatur löst sich der ausfallende Feststoff wieder. Die hellgelbe Lösung wird noch ca. 30 Minuten bei Raumtemperatur gerührt. Nach Abkühlen der Lösung auf -65°C wird langsam eine Lösung von 1.63 g (10 mmol) Ethan-1,2-disulfenyl Chlorid in 10 ml getrocknetem Diethylether zugetropft. Die Lösung wird langsam auf Raumtemperatur gebracht, wobei ein weißer Feststoff ausfällt. Der Reaktionsansatz wird über Nacht weitergerührt. Dann wird vorsichtig mit ca. 20 ml destilliertem Wasser hydrolisiert, eventuell nicht gelöster Feststoff abfiltriert (Produkt !), die Phasen getrennt und die wäßrige Phase zweimal mit ca. 20 ml Diethylether und zweimal mit Dichlormethan ausgeschüttelt. Die vereinigte organische Phase wird über Magnesiumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Der zurückbleibende weiße Feststoff wird in Pentan aufgenommen. Das Produkt kristallisiert bei -20 °C aus. Molmasse: 502.83 g mol-1 Ausbeute: 3.62g (72 %) C30H46O2S2 Elementaranalyse: C% H% S% berechnet: 71.66 9.22 12.75 gefunden: 72.11 9.43 12.41 Massenspektrum: 502 m/z [H2bpte]+ Kernresonanzspektren: 1 H-NMR (CDCl3, 400 MHz): δ 1.25 (18H, s); 1.37 (18H, s); 2.78 (4H, s); 7.03 (2H, s); 7.28 (2H, d); 7.30 (2H, d) ppm 13 C-NMR (CDCl3, 100 MHz): δ 29.42; 31.51; 34.29; 35.20; 36.24; 117.44; 126.10; 130.30; 135.31; 142.35;153.18 ppm Präparativer Teil 122 Darstellung von Cu2(bse)2: Zu einer Lösung von 251 mg (0.5 mmol) H2bse in 25 ml getrocknetem THF wird unter S Argon 49 mg (0.5 mmol) Cu(I)Cl S zugegeben, in der Siedehitze gelöst und 140 µl (1mmol) getrocknetes O Cu O O Cu O S S Triethylamin zugegeben. Nach einer Stunden kochen unter Rückfluß wird die Lösung auf Raumtemperatur abgekühlt und über Nacht bei –20 °C verwahrt. Über Nacht kristallisiert TEAHCl in weißen Nadeln aus. Dieses wird abfiltriert, und das Lösungsmittel am Rotationsverdampfer entfernt. Der zurückbleibende grüne Feststoff wird aus Aceton umkristallisiert. Ausbeute: 60 mg (21 %) Molmasse: 1128.71 g mol-1 C60H88S4O4Cu2 Elementaranalyse: C% H% S% berechnet: 63.85 7.86 11.36 gefunden: 63.85 7.99 11.08 Massenspektrum: 1128 m/z [Cu2(bse)2]+ IR-Spektrum: 80 75 70 T [% ] 65 60 55 50 45 40 4000 3500 3000 2500 2000 1500 -1 W ellenzahl [cm ] 1000 500 Präparativer Teil 123 Darstellung von Co(bsb)(Hbsb): Zu einer Lösung von 530 mg (1mmol) H2bsb in 40 ml getrocknetem THF wird unter Argon 130 mg (1 S S Co S Co(II)Cl2 zugegeben, in der OH S O Siedehitze gelöst und 280 µl (2 mmol) O mmol) O getrocknetes Triethylamin zugegeben. Nach einer Stunde kochen unter Rückfluß wird die Lösung unter Sauerstoffzufuhr auf Raumtemperatur abgekühlt. Die Farbe der Lösung schlägt hierbei von grün nach braun um. Über Nacht kristallisiert bei –20°C Triethylammoniumhydrochlorid in weißen Nadeln aus. Dieses wird abfiltriert und das Lösungsmittel am Rotationsverdampfer entfernt. Der zurückbleibende braune Feststoff wird aus Chloroform/Diethylether (1:1) umkristallisiert. Ausbeute: Molmasse: 1157.65 g C68H89S4O4Co 220 mg (38 %) Elementaranalyse: C% H% S% berechnet: 70.55 7.75 11.08 gefunden: 70.12 7.88 10.86 Massenspektrum: 1215 m/z [Co2(bsb)2]+ IR-Spektrum: 70 T [% ] 65 60 55 50 45 40 4000 3500 3000 2500 2000 1500 -1 W e lle n z a h l [c m ] 1000 500 Präparativer Teil 124 Darstellung von Cu2(bsb)2: Zu einer Lösung von 530 mg (1mmol) H2bsb in 40 ml getrocknetem THF wird unter S Argon 98 mg (1 mmol) Cu(I)Cl S zugegeben, in der Siedehitze gelöst und 280 µl (2 mmol) getrocknetes O Cu O O Cu O S S Triethylamin zugegeben. Nach einer Stunde kochen unter Rückfluß wird die Lösung auf Raumtemperatur abgekühlt. Über Nacht kristallisiert bei –20°C Triethylammoniumhydrochlorid in weißen Nadeln aus. Dieses wird abfiltriert und das Lösungsmittel am Rotationsverdampfer entfernt. Der zurückbleibende braune Feststoff wird aus Dichlormethan umkristallisiert. Ausbeute:180 mg (29 %) Molmasse: 1224.80 g mol–1 C68H88S4O4Cu2 Elementaranalyse: C% H% S% berechnet: 66.68 7.24 10.47 gefunden: 66.43 7.19 10.38 Massenspektrum: 1224 m/z [Cu2(bsb)2]+ T [%] IR-Spektrum: 80 78 76 74 72 70 68 66 64 62 4000 3500 3000 2500 2000 1500 -1 W ellenzahl [cm ] 1000 500 Präparativer Teil 125 Darstellung von Cu(bsb)(py)2: 612 mg (0.5 mmol) [Cu2(bsb)2] werden in 20 ml Dichlormethan gelöst und mit 79 mg S (1 mmol) Pyridin versetzt. Die Lösung ändert ihre Farbe dabei von braun nach schwach violett. Bei N Cu S langsamen O O N Abdampfen des Lösungsmittels erhält man den Komplex in Form schwach violetter Kristalle. Ausbeute:193 mg (26 %) Molmasse: 770.60 g mol-1 C44H54S2O2N2Cu Elementaranalyse: C% H% S% berechnet: 68.58 7.06 8.32 gefunden: 68.21 6.97 8.54 Massenspektrum: 611 m/z [Cu(bsb)]+ T [%] IR-Spektrum: 84 82 80 78 76 74 72 70 68 4000 3500 3000 2500 2000 1500 -1 W ellenzahl [cm ] 1000 500 Präparativer Teil 126 Darstellung von [Pt(psp)2]: Zu einer Lösung von 314 mg (1 mmol) Hpsp und 1 ml einer 1 molaren S Pt Natriummethanolatlösung in 30 ml getrocknetem Methanol wird S O O bei Raumtemperatur unter Argon 133 mg (0.5 mmol) PtCl2 zugegeben. Nach zwei Stunden kochen unter Rückfluß wird heiß filtriert und das Filtrat an Luft stehengelassen. Über Nacht kristallisiert der Komplex aus dem Filtrat in gelben Nadeln aus. Ausbeute:170 mg (41 %) Molmasse: 822.05 g mol-1 C40H50S2O2Pt Elementaranalyse: C% H% S% berechnet: 58.44 6.13 7.80 gefunden: 58.21 6.08 7.93 Massenspektrum: 821 m/z [Pt(psp)2]+ T [%] IR-Spektrum: 85 80 75 70 65 60 55 50 45 40 4000 3500 3000 2500 2000 1500 -1 Wellenzahl [cm ] 1000 500 Präparativer Teil 127 Darstellung von [Cu(psp)2]: Zu einer Lösung von 98 mg (1 mmol) S CuCl (wasserfrei) und 628 mg (2 O Cu mmol) Hpsp in 30 ml trockenem O S Methanol werden bei Raumtemperatur unter Argon trockenes 280 ml (2 mmol) Triethylamin zugetropft. Nach drei Stunden kochen unter Rückfluß wird filtriert und das Filtrat an Luft stehengelassen. Über Nacht kristallisiert der Komplex aus dem Filtrat in braunen Nadeln aus. Ausbeute:255 mg (37 %) Molmasse: 690.51 g mol-1 C40H50S2O2Cu Elementaranalyse: C% H% S% berechnet: 69.58 7.30 9.29 gefunden: 68.40 7.46 9.80 Massenspektrum: 689 m/z [Cu(psp)2]+ T [%] IR-Spektrum: 84 82 80 78 76 74 72 70 68 66 64 4000 3500 3000 2500 2000 1500 -1 Wellenzahl [cm ] 1000 500 Präparativer Teil 128 Darstellung von [Pd(psp)2]: Zu einer Lösung von 628 mg (2 mmol) Hpsp und 2 ml einer 1 molaren S S Natrium-methanolatlösung in 30 ml getrocknetem Methanol wird Pd O bei O Raumtemperatur unter Argon 177 mg (1 mmol) PdCl2 zugegeben. Nach zwei Stunden kochen unter Rückfluß wird der ausgefallene violette Niederschlag heiß abfiltriert. Dieser kann aus Hexan kristallisiert werden. Ausbeute:450 mg (61 %) Molmasse: 733.39 g mol-1 C40H50S2O2Pd Elementaranalyse: C% H% S% berechnet: 65.51 6.87 8.74 gefunden: 65.82 6.71 8.41 Massenspektrum: 732 m/z [Pd(psp)2]+ IR-Spektrum: 84 82 80 T [%] 78 76 74 72 70 4000 3500 3000 2500 2000 1500 -1 Wellenzahl [cm ] 1000 500 Präparativer Teil 129 Darstellung von [Cu(bsbu)]: 530 mg (1mmol) H2bsbu werden mit 2 ml S S 1 molarer methanolischer NaOH-Lösung Cu O unter Rückfluss in 40 ml getrocknetem O Methanol gelöst. Eine Lösung von 242 mg (1mmol) Cu(NO3)2 3 H2O in 4 ml getrocknetem Methanol wird in der Siedehitze zugegeben. Es fällt ein brauner Niederschlag. Nach einer Stunde Rühren wird der Niederschlag abfiltriert und aus Dichlormethan umkristallisiert. Ausbeute:190 mg (32 %) Molmasse: 592.41 g mol-1 C32H48S2O2Cu Elementaranalyse: berechnet: Massenspektrum: C% H% S% 64.88 8.17 10.83 591 m/z [Cu(bsbu)]+ IR-Spektrum: 80 T [%] 70 60 50 40 30 4000 3500 3000 2500 2000 1500 -1 W ellenzahl [cm ] 1000 500 Präparativer Teil 130 Darstellung von [Pt(bsbu)]: 530 mg (1mmol) H2bsbu werden mit 280 S S Pt ml (2 mmol) getrocknetem Triethylamin in O 30 ml getrocknetem Acetonitril vorgelegt O und bei Raumtemperatur 266 mg (1 mmol) PtCl2 zugegeben. Die resultierende grün-braune Suspension wird zum Rückfluss erhitzt, wobei eine gelbe Lösung entsteht. Nach einer Stunde rühren unter Rückfluss wird die Lösung auf Raumtemperatur abgekühlt, wobei ein hellgelber Niederschlag ausfällt. Dieser wird abfiltriert und mit Acetonitril und Methanol gewaschen. Ausbeute:320 mg (44 %) Molmasse: 723.94 g mol-1 C32H48S2O2Pt Elementaranalyse: berechnet: Massenspektrum: C% H% S% 53.09 6.68 8.86 723 m/z [Pt(bsbu)]+ IR-Spektrum: 85 80 T [%] 75 70 65 60 55 50 4000 3500 3000 2500 2000 1500 -1 W ellenzahl [cm ] 1000 500 Präparativer Teil 131 Darstellung von [Pt(bsbu)]: 265 mg (0.5mmol) H2bsbu werden mit 140 ml (1 mmol) getrocknetem Pd O Triethylamin in 30 ml getrocknetem Acetonitril vorgelegt und S S O bei Raumtemperatur 89 mg (0.5 mmol) PdCl2 zugegeben. Nach einer Stunde rühren unter Rückfluss wird die Lösung auf Raumtemperatur abgekühlt, wobei ein rötlicher Niederschlag ausfällt. Dieser wird abfiltriert und mit Acetonitril und Methanol gewaschen. Ausbeute:92 mg (29 %) Molmasse: 635.28 g mol-1 C32H48S2O2Pd Elementaranalyse: berechnet: Massenspektrum: C% H% S% 60.50 7.62 10.10 634 m/z [Pd(bsbu)]+ IR-Spektrum: 80 T [%] 70 60 50 40 4000 3500 3000 2500 2000 1500 -1 W ellenzahl [cm ] 1000 500 Anhang 11. 132 Anhang Cis-Trans-Isomerisierung des Komplexes [Pd(psp)2] 90 80 70 % trans 60 50 40 30 20 10 -200 0 200 400 600 800 1000 1200 1400 1600 1800 t [min] 243 K 87 86 % trans 85 84 83 82 81 80 0 50 100 150 t [min] 263 K 200 250 Anhang 133 80 70 % trans 60 50 40 30 20 0 20 40 60 80 100 120 140 160 180 200 t [m in] 273 K 92 90 88 % cis 86 84 82 80 78 76 0 20 40 60 t [min] 283 K 80 100 Anhang 134 Kristalldaten Ligand H2bse Hpsp Lösungsmittel Tetrahydrofuran Methanol Summenformel C30 H46 O2 S2 C20 H26 O S Molekulargewicht / g mol-1 502.79 314.47 Kristallabmessungen / mm 0.52 x 0.42 x 0.12 0.40 x 0.40 x 0.30 1.153 1.136 Kristallsystem Monoklin Monoklin Raumgruppe P21/c P21/c a/Å 16.148 (3) 8.619 (2) b/Å 10.006 (2) 12.585 (3) c/Å 9.060 (2) 17.367 (4) α/° 90 90 β/° 98.44 (3) 102.55 (3) γ/° 90 90 Volumen / Å3 1448.0 (5) 1838 (6) Z 2 4 Absorbtionskoeffizient µ / mm 0.208 0.176 Diffraktometer Siemens SMART Siemens SMART Strahlung Mo-Kα Mo-Kα Temperatur / K 100 (2) 100 (2) Meßbereich 2υ / ° 2.40 bis 25.00 2.02 bis 30.00 gemessene Reflexe 10771 17296 unabhängige Reflexe 2527 5258 verfeierte Parameter 548 680 R-Wert / % 0.0822 0.1060 RW-Wert 0.1683 0.2375 GOOF 1.094 1.099 Restelektronendichte / e Å-3 0.432 und –0.439 0.878 und –0.534 Berechnete Dichte / g cm -3 Messdaten Anhang 135 Kristalldaten Ligand H2bsb Lösungsmittel Aceton Summenformel C34 H46 O2 S2 Molekulargewicht / g mol-1 550.83 Kristallabmessungen / mm 0.2 x 0.2 x 0.2 Berechnete Dichte / g cm-3 1.141 Kristallsystem Monoklin Raumgruppe P21/n a/Å 16.175 (3) b/Å 12.580 (3) c/Å 17.102 (3) α/° 90 β/° 112.88 (3) γ/° 90 Volumen / Å3 3206.1 (11) Z 4 Absorbtionskoeffizient µ / mm 0.193 Messdaten Diffraktometer Siemens SMART Strahlung Mo-Kα Temperatur / K 100 (2) Meßbereich 2υ / ° 2.07 bis 24.99 gemessene Reflexe 12814 unabhängige Reflexe 5186 verfeierte Parameter 1192 R-Wert / % 0.0796 RW-Wert 0.1105 GOOF 0.930 Restelektronendichte / e Å-3 0.338 und –0.226 Anhang 136 Kristalldaten Komplex Cu2(bse)2 Cu2(bsb)2 Lösungsmittel Aceton Dichlormethan Summenformel C60 H88 Cu2 O4 S4 C68 H88 Cu2 O4 S4 Molekulargewicht / g mol-1 1128.62 1224.70 Kristallabmessungen / mm 0.26 x 0.24 x 0.06 0.60 x 0.07 x 0.06 Berechnete Dichte / g cm-3 1.242 1.228 Kristallsystem Monoklin Monoklin Raumgruppe C2/c P21/c a/Å 19.145 (2) 12.384 (3) b/Å 10.486 (1) 15.281 (3) c/Å 31.250 (3) 17.624 (4) α/° 90 90 β/° 105.83 (2) 94.06 (3) γ/° 90 90 Volumen / Å3 6035.8 (10) 3313.1 (13) Z 4 2 Absorbtionskoeffizient µ / mm 0.886 0.812 Diffraktometer Siemens SMART Nonius Kappa-CCD Strahlung Mo-Kα Mo-Kα Temperatur / K 100 (2) 100 (2) Meßbereich 2υ / ° 2.21 bis 23.26 2.12 bis 22.50 gemessene Reflexe 20023 14140 unabhängige Reflexe 4333 4331 verfeierte Parameter 2408 1300 R-Wert / % 0.0942 0.1266 RW-Wert 0.0865 0.2073 GOOF 0.971 1.072 Restelektronendichte / e Å-3 0.279 und –0.431 0.839 und –0.464 Messdaten Anhang 137 Kristalldaten Komplex Cu(bsb)(py)2 Co(bsb)(Hbsb) Lösungsmittel Dichlormethan Chloroform Summenformel C44 H54 Cu2 N2 O2 S2 C68 H89 Co O4 S4 *1.5CHCl3*0.5 H2O Molekulargewicht / g mol-1 770.55 1345.70 Kristallabmessungen / mm 0.14 x 0.11 x 0.07 0.30 x 0.12 x 0.06 1.262 1.210 Kristallsystem Monoklin Monoklin Raumgruppe P21/n P21/n a/Å 9.804 (2) 20.122 (2) b/Å 18.357 (4) 16.202 (2) c/Å 22.535 (5) 24.477 (3) α/° 90 90 β/° 90.76 (2) 112.22 (2) γ/° 90 90 Volumen / Å3 4055 (2) 7387 (2) Z 4 4 Absorbtionskoeffizient µ / mm 0.679 0.552 Berechnete Dichte / g cm -3 Messdaten Diffraktometer Nonius Kappa-CCD Nonius Kappa-CCD Strahlung Mo-Kα Mo-Kα Temperatur / K 100 (2) 100 (2) Meßbereich 2υ / ° 1.81 bis 22.50 2.19 bis 25.00 gemessene Reflexe 13568 31999 unabhängige Reflexe 5288 12898 verfeierte Parameter 1636 2848 R-Wert / % 0.1269 0.1365 RW-Wert 0.1074 0.3102 GOOF 0.963 1.065 Restelektronendichte / e Å-3 0.333 und –0.328 1.714 und –0.724 Anhang 138 Kristalldaten Komplex Cu(psp)2 Pt(psp)2 Lösungsmittel Methanol Methanol Summenformel C40 H50 Cu O2 S2 C40 H50 Pt O2 S2 Molekulargewicht / g mol-1 690.46 822.01 Kristallabmessungen / mm 0.28 x 0.18 x 0.06 0.55 x 0.12 x 0.07 Berechnete Dichte / g cm-3 1.237 1.460 Kristallsystem Monoklin Monoklin Raumgruppe C2/c P21/c a/Å 29.729 (4) 12.9473 (9) b/Å 13.875 (2) 11.1651 (9) c/Å 21.983 (3) 26.008 (2) α/° 90 90 β/° 125.12 (2) 95.83 (2) γ/° 90 90 Volumen / Å3 7417 (2) 3740.2 (5) Z 8 4 Absorbtionskoeffizient µ / mm 0.733 3.895 Messdaten Diffraktometer Nonius Kappa-CCD Siemens SMART Strahlung Mo-Kα Mo-Kα Temperatur / K 100 (2) 100 (2) Meßbereich 2υ / ° 2.55 bis 27.50 1.57 bis 27.50 gemessene Reflexe 54186 31815 unabhängige Reflexe 8520 8553 verfeierte Parameter 2936 1664 R-Wert / % 0.0897 0.0705 RW-Wert 0.1635 0.1215 GOOF 1.049 1.016 Restelektronendichte / e Å-3 1.313 und –0.539 3.482 und –2.114 Anhang 139 Kristalldaten Komplex Pd(psp)2 Lösungsmittel Hexan Summenformel C40 H50 Pd O2 S2 Molekulargewicht / g mol-1 733.32 Kristallabmessungen / mm 0.38 x 0.26 x 0.04 Berechnete Dichte / g cm-3 1.323 Kristallsystem Triklin Raumgruppe P-1 a/Å 14.203 (1) b/Å 14.836 (1) c/Å 14.914 (1) α/° 110.29 (2) β/° 108.70 (2) γ/° 91.13 (2) Volumen / Å3 2761.5 (3) Z 3 Absorbtionskoeffizient µ / mm 0.650 Messdaten Diffraktometer Siemens SMART Strahlung Mo-Kα Temperatur / K 100 (2) Meßbereich 2υ / ° 1.53 bis 24.96 gemessene Reflexe 11747 unabhängige Reflexe 8645 verfeierte Parameter 1152 R-Wert / % 0.0908 RW-Wert 0.0989 GOOF 0.946 Restelektronendichte / e Å-3 0.537 und –0.731 Literaturverzeichnis 11. [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] [22] 140 Literaturverzeichnis G. T. Babcock, B. A. Barry, R. J. Debus, C. W: Hoganson, M. Atamian, L. McIntosh, I. Sithole, C. F. Yokum Biochemistry 1989, 28, 9557 P. Nordlund, B.-M. Sjöberg, H. Eklund Nature 1990, 345, 593 G. T. Babcock, M. K. El-Deeb, P. O. Sandusky, M. M. Whittaker, J. W. Whittaker J. Am. Chem. Soc. 1992, 114, 3727 M. M: Whittaker, P. J. Kersten, N. Nakamura, J. Sandes-Loehr, E. S. Schweizer, J. W. Whittaker J. Biol. Chem. 1996, 271, 681 M. Fontecave, J.-L. Pierre Bull. Soc. Chim. Fr. 1996, 133, 653 J. A. D. Cooper, W. Smith, M. Bacila, H. Medina J. Biol. Chem. 1959, 234, 445-448 N. Ito, S. E.V. Phillips, C. Stevens, Z. B. Ogel, M. J. McPherson, J. N. Keen, K. D. S. Yadav, P. F. Knowels Nature 1991, 350, 87 J. A. Halfen, B. A. Jazdzewski, S. Mahapatra, L. M. Berreau, E. C. Wikinson, L. Que, W. B. Tolman J. Am. Chem. Soc. 1997, 119, 8217 F. Himo, G. T. Babcock, L. A. Eriksson Chem. Phys. Lett. 1999, 313, 374 E. W. Wise, J. B. Pate, R. A. Wheeler J. Phys. Chem. B 1999, 103, 4772 F. Himo, L. A. Eriksson, M. R. A. Blomberg, P. E. M. Siegbahn Int. J. Quantum Chem. 2000, 76, 714 F. Himo, L. A. Eriksson, F. Maseras, P. E. M. Siegbahn J. Am. Chem. Soc. 2000, 122, 8031-36 U. Wallmann Dissertation Ruhr-Universität Bochum 1998 A. Sokolowski, E. Bothe, E. Bill, T. Weyhermüller, K. Wieghardt Chem. Comm. 1996, 1671 A. Sokolowski, J. Müller, T. Weyhermüller, R. Schnepf, P. Hildebrandt, K. Hindenbrand, E. Bothe, K. Wieghardt J. Am. Chem. Soc. 1997, 119, 8889 B. Adams, E. Bill, E. Bothe, B. Goerdt, G. Haselhorst, K. Hildenbrandt, A. Sokolowski, S. Steenken, T. Weyhermüller, K. Wieghardt Chem. Eur. J. 1997, 3, 308 M. D. Snodin, L. Ould-Moussaa, U. Wallmann, S. Lecomte, V. Bachler, E. Bill, H. Hummel, T. Weyhermüller, P. Hildebrandt, K. Wieghardt Chem. Eur. J. 1999, 5, 2554 R. Schnepf, A. Sokolowski, J. Müller, V. Bachler, K. Wieghardt, P. Hildebrandt J. Am. Chem. Soc. 1998, 120, 2352 D. Zurita, C. Scheer, J. L. Pierre, E. Saint-Aman J. Chem. Soc., Dalton Trans. 1996, 4331 D. Zurita, I. Gautier-Luneau, S. Menage, J.-L. Pierre, E. SaintAman J. Biol. Inorg. Chem. 1997, 2, 46 D. Zurita, S. Menage, J.-L. Pierre, E. Saint-Aman New J. Chem. 1997, 21, 1001 J. Müller, T. Weyhermüller, E. Bill, P. Hildebrandt, L. OuldMoussa, T. Glaser, K. Wieghardt Angew. Chem. 1998, 110, 637 Literaturverzeichnis [23] [24] [25] [26] [27] [28] [29] [30] [31] [32] [33] [34] [35] [36] [37] [38] [39] [40] [41] [42] [43] [44] [45] [46] [47] [48] [49] [50] 141 A. Sokolowski Dissertation Ruhr-Universität Bochum 1996 F. N. Penkert Dissertation Ruhr-Universität Bochum 2000 S. Kimura Dissertation Ruhr-Universität Bochum 2000 Y. Wang, T. D. P. Stack J. Am. Chem. Soc. 1996, 118, 13097 E. Saint-Aman, S. Ménage, J. L. Pierre, E. Defrancq, G. Gellon New J. Chem. 1998, 37, 2217 P. Chaudhuri, M. Hess, T. Weyhermüller, K. Wieghardt Angew. Chem., Int. Ed. 1998, 38, 1095 P. Chaudhuri, M. Hess, J. Müller, K. Hildenbrandt, E. Bill, T. Weyhermüller, K. Wieghardt J. Am. Chem. Soc. 1999, 121,9599 P. Chaudhuri, M. Hess, U. Flörke, K. Wieghardt Angew.Chem., Int. Ed. 1998, 37, 2217 I. W. J. Still, G. W. Kutney, D. McLean J. Org. Chem. 1982, 47, 560-561 W. H. Mueller, M. Dines J. Heterocycl. Chem. 1969, 6, 627-630 H. Brintzinger M. Langheck, H. Ellwanger Chem. Ber. 1954, 87, 320-324 O. Kahn, Inorg. Chim. Acta 1982, 62, 3-14 V. H. Crawford, H.W. Richardson, J.R. Wasson, D. J. Hodgson, W. E. Hatfield Inorg. Chem. 1976, 15, 2107-2110 M. Melnik Coord. Chem.Rev. 1982, 42, 259-293 S. K. Mandal, L. K. Thompson, K. Nag, J.-P. Charland, E. J. Gabe Can. J. Chem. 1987, 65, 2815-2823 O. Kahn Angew. Chem. 1985, 24, 834 W. Mazurek, B. J. Kennedy, K. S. Murray, M.J. O´Connor, J. R. Rodgers, M. R. Snow, A. G. Wedd, P. R. Zwack Inorg. Chem. 1985, 24, 3258-3264 J. Glerup, D. J. Hodgson, E. Pedersen Acta Chem. Scand. Ser. A 1983, A37, 161 S. Kallasoe, E. Pederson. Acta Chem. Scand. Ser. 1982, A36, 859 J. Bertrand, C. E. Kirkwood Inorg. Chim. Acta 1972, 6, 248 A. Bensini, D. Gatteschi Inorg. Chim. Acta. 1978, 31, 11-18 A. Sokolowski, H. Leutbecher, T. Weyhermüller, R. Schnepf, E. Bothe, E. Bill, P. Hildebrandt, K. Wieghardt J. Biol. Inorg. Chem. 1997, 2, 444-53 M. A. Halcrow, L. M. L. Chia, X. Liu, E. J. L. McInnes, L. J. Yellowless, F. E. Mabbs, I. J. Scowen, M. McPartlin, J. E. Davies Inorg. Chem. 1999, 1753-62 S. Itoh, S. Takayama, R. Arakawa, A. Furuta, M. Komatsu, A. Ishida, S. Takamuk, S. Fukuzumi Inorg. Chem. 1997, 36,140716 M. Taki, H. Kumai, S. Nagatomo, T. Kitagawa, S. Itho, S. Fukuzumi, Inorg. Chim. Acta 2000, 300-302, 622-32 M. M. Whittaker, W. R. Duncan, J. W. Whittaker, Inorg. Chem. 1996, 35, 382-86 S. Itoh, M. Taki, S. Fukuzumi, Coord. Chem. Rev. 2000, 198, 320 Y. Shimazaki, S. Huth, S. Hirota, O. Yamauchi, Bull. Chem. Soc. Jpn. 2000, 73, 1187-95 Literaturverzeichnis [51] [52] [53] [54] [55] [56] [57] [58] [59] [60] [61] [62] [63] [64] [65] [66] [67] [68] [69] [70] [71] [72] [73] [74] [75] 142 M. Veidis J. Am. Chem. Soc. 1969, 91, 1859 P. Chaudhuri, K. Oder, K. Wieghardt, J. Weiss, J. Reedijk, W. Hinrichs, J. Wood, A. Ozarowski, H. Stratemaier, D. Reinen Inorg. Chem. 1986, 25, 2951 A. D. Beveridge, A. J. Lavery, M.D. Walkinshaw, M. Schröder J. Chem. Soc. 1987, 373 J. Müller Dissertation Ruhr-Universität Bochum 1999 A. Sokolowski, B. Adam, T. Weyhermüller, A. Kikuchi, K. Hildenbrand, R. Schnepf, P. Hildebrandt, E. Bill, K. Wieghardt Inorg. Chem. 1997, 36, 3702-10 T. M. Donlevy, L. R. Gahan, T. W. Hambley, R. Strangler Inorg. Chem. 1992, 31, 4376-82 T. W. Hambley, L. R. Gahan, G. H. Searle Acta Crystallogr., Sect. C 1989, C45, 864 T. W. Hambley Acta Crystallogr., Sect. C 1988, B44, 601 E. Bouwman, W. L. Driessen J. Am. Chem. Soc. 1988, 110, 4440-1 L. Roecker, M. H. Dickman, D. L. Nosco, R. J. Doedens, E. Deutsch Inorg. Chem. 1983, 22, 2022-28 A. B. P. Lever Inorganic Electronic Spektroskopy 2. Auflage, Elsevier, Amsterdam 1984 G. Gerfen, B. F. Bellew, R. G. Griffen, D. J. Singel, C. A. Ekberg, J. W. Whittacker J. Phys. Chem. 1997, 100, 16739 M. M. Whittaker, C. A. Ekberg, J. Peterson, M. S. Sendova, E. P. Day, J. W. Whittaker J. Mol. Cat. B: Enzymatic 2000, 8, 3-15 S. Itho, M. Taki, S. TAkayama, S. Nagatomo, T. Kitagawa, N. Sakurada, R. Arakawa, S. Fukuzumi Angew. Chem. 1999, 111, 2944-46 M. M. Whittaker, Y.-Y. Chuang, J. W. Whittaker J. Am. Chem. Soc. 1993, 115, 10029-10035 M. M. Whittaker, D. B. Ballou, J. W. Whittaker Biochemistry 1998, 37, 8426-8436 J. Müller, A. Kikuchi, E. Bill, T. Weyhermüller, P. Hildebrandt, L. Ould-Moussa, K. Wieghardt Inorg. Chim. Acta 2000, 297, 265-277 R. M. Buchanan, C. G. Pierpont J. Am. Chem. Soc. 1980, 102, 4951 P. A. Wicklund, L. S. Beckmann, D. G. Brown Inorg. Chem. 1976, 15, 1996 S. L. Kessel, R. M. Emberson, P. Debrunner, D. N. Hendrickson, Inorg. Chem. 1980, 19, 1170 C. W. Lange, B. J. Conklin, C. G. Pierpont Inorg. Chem. 1994, 33, 1276 M. Hesse, H. Meier, B. Zeeh, Spektroskopische Methoden in der organischen Chemie, 5. Auflage, Georg Thieme Verlag, Stuttgart, 1995 E. W. Abel, G. W. Farrow, K. G. Orrell, V. Sik J. Chem. Soc. Dalton Trans. 1977, 42-46 P. C. Turley, P. Haake J. Am. Chem. Soc. 1967, 4617-21 R. T. Boeré, J. Willis Inorg. Chem. 1985, 24, 1059-65 Literaturverzeichnis [76] [77] [78] [79] [80] [81] [82] [83] [84] [85] [86] [87] [88] [89] [90] [91] [92] [93] [94] [95] [96] 143 R. T. Boeré, J. Willis Can. J. Chem. 1985, 63, 3530 A. Sokolowski Dissertation Ruhr-Universität Bochum 1996 M. Born Z. Phys. 1920, 1920, 1, 45 J. O´M. Brockris, A. K. N. Reddy Modern Elektrochemistry, Vol. 1, Plenum Press, New York, 1970 G. N. R. Tripathi, Q. Sun, D. A. Armstrong, D. M. Chipman, R. H. Schuler J. Phys. Chem. 1992, 96, 5344-50 M. Bonifacic, J. Weiss, S. A. Chaudhri, K.-D. Asmus J. Phys. Chem. 1985, 89, 3910-3914 M. Geoffroy, G. Bernardinelli, P. Castan, H. Chermette, D. Deguenon, S. Nour, J. Weber, M. Wermeille Inorg. Chem. 1992, 31, 5056-5060 R. T. Boeré, C. J. Willis Inorg. Chem. 1985, 24, 1065-1069 S. R. Logan, Grundlagen der chemischen Kinetik, Wiley-VCH Verlag GmbH Weinheim, 1985 F. Basolo, R. G. Pearson Mechanismen in der anorganischen Chemie, Georg Thieme Verlag, Stuttgart, 1973 R. J. Cross Chem. Soc. Rev. 1985, 14, 197 R. J. Cross Adv. Inorg. Chem. 1989, 34, 219 L. Cattalini, Hrsg. J. O. Edwards, Inorganic Reaction Mechanism, Wiley-Interscience, New York, 1970 R. G. Pearson Inorg. Chem. 1973, 12, 712-713 E. J. Little, M. M. Jones J. Chem. Ed. 1960, 37, 231 J. M. Lambert Top. Stereochem. 1971, 6, 19 M. G. Patch, K. P. Simolo, C. J. Carrano Inorg. Chem. 1982, 21, 2972 F. Neese, unveröffentlicht, Computerprogramm „EPR“ Version 1.0, 1994 E. Bill, unveröffentlicht, Programmpacket: ESIM B. J. Gaffney, J. Silverstone in Biological Magnetic Resonanz, Vol. 13, EMR of Paramagnetic Molecules (Hrsg. L. J. Berliner, J. Reuben), Plenum Press, New York, 1993 W. H. Press, B. P. Flannery, S. A. Tukolsky, W. T. Vutterly Numerical Recipes Cambridge University Press, Cambridge, 1990 Lebenslauf Name Tobias Kruse Geburtsdatum 08.11.1970 Geburtsort Greven Familienstand ledig Schulausbildung 1977-1981 Grundschule Steinfurt 1981-1990 Gymnasium Steinfurt Mai 1990 allgemeine Hochschulreife Wehrdienst Juli 1990 bis Grundwehrdienst Juni 1991 in Wuppertal und Münster Studium Oktober 1991 Beginn des Diplomstudiengangs Chemie an der Heinrich-Heine-Universität Düsseldorf Februar 1994 Diplom-Vorprüfung in Chemie Mai 1997 Diplom-Hauptprüfung in Chemie August 1997 bis Anfertigung der Diplomarbeit am Lehrstuhl I Februar 1998 des Institutes für Organische und Makromolekulare Chemie (Prof. H.-D. Martin) März 1998 bis Anfertigung der Dissertation am Max-Planck- Februar 2001 Institut für Strahlenchemie in Mülheim an der Ruhr (Prof. K. Wieghardt) Berufstätigkeit März 1998 bis Stipendiat am Max-Planck-Institut für Strahlen- Februar 2001 chemie in Mülheim an der Ruhr